Avogadro Number’s-oriented HyperGeometric and ChebyshevT Functions for Black Hole Paradox Generalizations and Turing Machine Ruled Quantum Homeopathy Water Memory Entanglements for the Translation of COVID19
Avogadro Number’s-oriented HyperGeometric and ChebyshevT Functions for Black Hole Paradox Generalizations and Turing Machine Ruled Quantum Homeopathy Water Memory Entanglements for the Translation of COVID19 Homeopathy Remedies into the Neprilysin and ACE2/AT1R Receptors Targeted DRVYIHPFX- Ligands.
Grigoriadis Ioannis1*,2,3
1Department of Biogenetoligandorol QMMIDDD/QPRPICA/MACHNOT/QIICDNNDCA ADMET/QIICDNNDCA Stations.
2Cell-Pharmacy Ltd, Personalized Synthocure TM Stations.
3Biogenea Pharmaceuticals Ltd
*Corresponding Author:Grigoriadis Ioannis, Biogenea Pharmaceuticals TM, Thessaloniki, 26th October Str, no: 43, LIMANI CENTER, 5th Floor - 5.06, 546 27, Bilkent University, Turkey, Tel: 2310 254115-7, Fax: 2310 282867, E-mail:
Citation: Grigoriadis Ioannis (2024) Avogadro Number’s-oriented Hypergeometric and ChebyshevT Functions for Black Hole Paradox Generalizations and Turing Machine Ruled Quantum Homeopathy Water Memory Entanglements for the Translation of COVID19 Homeopathy Remedies into the Neprilysin and ACE2/AT1R Receptors Targeted DRVYIHPFX- Ligands. Medcina Intern 6: 216.
Received: February 25, 2024; Accepted: March 19, 2024; Published: March 23, 2024.
Copyright: © 2024 Grigoriadis Ioannis, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
SARS-CoV-2 Omicron variant spike RBD epitopes in complex with spike (S) -protein D614G mutations now predominate globally and increase infectivity by assembling more functional S proteins into the virion. On average, homeopathic medicine contains less than one molecule per dose on average. Single variable SphericalHarmonicY, LaguerreL, WhittakerM, SphericalBesselJ, LegendreP, LegendreQ, LaguerreL, ChebyshevT, and Hypergeometric1F1 Functions pFqarise in connection with the power series solution of the Schrodinger equation or in the summation of perturbation expansions in quantum mechanics. A quantum interpretation of the homeopathic method is presented here through a novel algebraic topology approach to supersymmetry and symmetry breaking in quantum fields and quantum gravity theory with the aim of developing a wide range of physical-based drug design applications in a Small Molecule Quantized Water Memory Network suggesting that the Hidden Black Hole Paradox of Information Loss might be solved under suitable conditions. Fascinating, although this water quantum memory mimicking topic is exploring the relationship between AdS5 Quantum fields Theory (QFT) Reduction, Vaidya and Kerr metrics for QFT to QM Reduction, Quantum Thermodynamics, and Turing Machine Learning Rules for Quantum Homeopathy Variables and Quantum Biology gives us a greater insight into viral transmissions and thus, SARS-CoV-2 biological complexities?

Math1a and

Keywords
Keywords: COVID19; SARS-COV-2 SPIKE D614G, Chern-Simons Topological; QED (Quantum Electrodynamics), Homeopathic Medicine; Water Memory; AI-Quantum Computing; Quantum-Inspired Evolutionary Algorithm Predictive Toxicology; QSAR Quantum Gates; CS Supergravity Quantum Foam; Cheminformatics Artificial Intelligence; Phase Data Mining; Machine Learning; Euclid Chemical Space Exploration; (Bosonic) Quantum Fields Theory (QFT); Angiotensin Receptor Neprilysin Targeted DRVYIHPFX Mimetic Holomorphic Ligands; Hidden Entanglement Negativity Translations; Uncertainty Quantum Relationships; Quantum Fields Theory; Quantum Information; Hypergeometric Functions; Chemical Block Systems; Black Hole Paradox Generalizations; Sphericalharmonicy-Supersymmetry Breaking Foundations; Sphericalharmonicy, Laguerrel, Hypergeometric1F1, Whittakerm Functions; Sphericalbesselj, Legendrep, Legendreq, Laguerrel, Chebyshevt, Hypergeometric1F1 Functions; Turing Machine Learning Ruled Calculations; Avogadro Number’s Oriented Quantum Homeopathy Hidden Entanglement Negativities; Quantum Foam; Non-Commutative Geometry; Small Molecule Ligand Engineering; Protein-Folding, Entropy, Enthalpy, H-Bonds, Thermodynamic; Extended Quantum Symmetries; Groupoids And Algebroid; Lie Algebras; Compact Quantum Groupoids; Quantum C ∗ -Algebras; Relativistic Quantum Gravity (RQG); Vaida Supergravity And Supersymmetry Theories; Hamiltonians in Quantum Gravity.
Summary
Computer-aided drug discovery/design methods which are broadly classified as either structure-based or ligand-based methods have played a major role in the development of therapeutically important small molecules for over three decades. In this strategy, an innovative Quantum Homeopathy-driven ligand replacement method translated here into a Structure-based evolutionary method which is in principle analogous to high-throughput screening for delocalizing pharmacophoric reconstructions that are perfectly well substituted in a fuzzy sphere shaped druggable scaffolding via an Avogadro Number’s-oriented quantum geometry and crowding-based low mass phase protocol for a phenotypic steady-state multiple genetic parental algorithm as a Quantum Geometry-based Dynamic Homeopathic Solution and for a Modified and Restricted Ligand Selection (QuGeDoHoSoMorLiP) approach. It also visualizes an idea that has been invoked in the description of other coherent energy transportations in organic molecules in the purpose of a better Turing Machine learning exploration of the energetic hypergeometric hypersurface for theidentification of multiple Quantum Phase Minima Homeopath Solutions in a single Hadamard run while preserving the chemical diversity of the generated RoccustyrnaTM structures. This could be a useful mathematical path for linking Quantum Negative Energy Harnessing from COVID19 Homeopathy remedies with Deep Geometry Sensing as described here which can be Turing Machine translated into Quantized Miasm Moiety metrics that exposed to the water interfaces and has sufficient electronic density data for the same purpose, for the designing of new crystallographic structures. In this case and in addition, important tools such as target/ligand data bases, homology modeling, ligand fingerprint methods, etc., necessary for successful implementation of various computer-aided drug discovery/design methods in this drug discovery campaign are discussed. The article combines quantum field theory with quantum homeopathy entanglement behind these most important Avogadro Number’s-oriented HyperGeometric and ChebyshevT Functions for Black Hole Paradox Generalizations where the substances which are not present, that is the homeopathic remedy, could become a kind of receptacle which ‘absorbs’ the Quantum Information extracted from COVID19 patient’s symptoms, insofar as they are related by similarity of these Turing Machine generated patterns/features, in a Gegenbauerian model which reconfirms the feasibility of our original intention for finding a new model-building recipe in this direction, a prototype Quantum entropy circuit that clearly illustrates a Quantum Homeopathy oriented design principle for a coherent exchange of single energy quanta between electronic and vibrational degrees of freedom with the capacity of calculating the enhanced negative free energy scoring functions from the best-docked poses of the Roccustyrna ligand.
Introduction
In light of the limited treatment modalities offered by conventional medicine for COVID-19, especially in more severe diseases, the results of the described homeopathic treatment in the cases previously presented are impressive because homeopathy is a well-established form of complementary and traditional medicine that has been used to treat a wide range of conditions, including respiratory illnesses [1,2,3,4,5]. Given the global impact of COVID-19 and despite the lack of a unified methodological standard, a number of healthcare organizations and ministries of health from various countries have recommended homeopathic treatments for the prevention and treatment of SARS-CoV-2 infections [1-4,5,6,7]. Inhibition of the SARS-CoV-2 Omicron variant spike RBD in complex with Fab XGv282 (PDB: 7wlc) binding interface interaction using small molecules or peptides is the most logical and straightforward strategy to block viral cellular entry. Consequently, our research team is committed to generating extended symmetries and quantum groupoid, algebroid, and functorial Avogadro Number’s representations demonstrating the efficacy of translating individualized homeopathy remedies in treating COVID-19 since homeopathic treatments have been employed successfully during major epidemics and pandemics of the 19th century, prompting numerous researchers to investigate the potential of homeopathic interventions to reduce the likelihood of SARS-CoV-2 infection or to alleviate COVID-19 symptoms [1-4,5-7,9,10]. Savera et al. 9 and Manchanda et al. reported that the most commonly prescribed homeopathic remedies for COVID-19 were Bryonia alba, Phosphorus, Arsenic album, Gelsemium sempervirens, and (Carboneum oxygenisatum or Pulsatilla nigricans). Clapers et al. conducted a prospective case series study of 103 mild-to-moderately ill COVID-19 patients in Spain, of whom 22 had concomitant diseases. The most frequently prescribed drugs were Bry, Ars, Phos, and Gels, whereas the drugs with the highest rate of” good response” were Sulphur (6/6 = 100%), Pulsatilla (4/5 = 85%), and Bryonia alba (21/29 = 72%) [1-9,10,11,12,13]. The time to complete recovery after homeopathic treatment ranged from 3.5 to 14.4 days, and potency 200c achieved the fastest rates of complete recovery and the least need to change the remedy. [1-9,10,11,12,13,14] Finally, very low response rates of 20% and 0% were observed with the use of Gelsemium and China officinalis, respectively, despite the broad recommendation emphasized in several studies. [9-14,15,16] Other controlled randomized studies corroborated these encouraging early findings and facilitated evidence-based decision-making regarding the role of homeopathy in treating COVID-19, adding a potentially quick and effective treatment modality that is both safe and inexpensive. [1-9,10,11,12,13,17,18] Pre-hypertension (ICD-10-CM R03.0) is a sub-clinical condition but remains a public health challenge globally. Appropriate intervention is needed to stop its progression to hypertension and other cardiovascular diseases. According to 8th Joint National Committee (JNC-8), blood pressure (BP) is classified as normotensive (≤ 120/80 mm Hg), high normal blood pressure or pre-hypertensive (120–139/80–89 mm Hg) and hypertensive (≥ 140/90 mm Hg). [1-9,10,11,12,13,19,20] Recent study has shown that during 2011–2012, 28.2% of US adults (≥ 20 years old) were pre-hypertensive. In 2014, a study in West Bengal police personnel showed that around 42.9% of police from various districts of West Bengal have pre-hypertension with increased risk of cardiovascular disease conditions. [1-9,10,11,12,13-20,21] Another study showed that pre-hypertension was present in 41.1% of Indian urban population of Belgaum city and recognized the necessity for detecting pre-hypertension and emphasizing on dietary and lifestyle modification (LSM) to prevent further progression to hypertension and its complications. [1-9,10,11,12-21] Controlling pre-hypertension is basically a primary prevention managed with diet, exercise, and lifestyle modifications. Now-a-days, complementary and alternative medicine (CAM) therapies are increasingly becoming popular in various chronic diseases including hypertension. Selective and integrative use of different CAM therapies has also been recommended in hypertension. Hypertension is a frequently encountered condition in routine homeopathy practice as well. Literature search revealed 13 published trials of homeopathy in hypertension. However, none on pre-hypertension. In the other trials, they hypothesized that there might be a significant difference between individualized homeopathic medicines (IH) and placebo in intervening with the progression of pre-hypertension to hypertension over 3 months of intervention while both groups received standardized recommendations for dietary and lifestyle modifications (LSM). [1-9,10-23] Recent calorimetric studies of small molecule interactions with biomolecular targets have generated renewed interest in the phenomenon of entropy-enthalpy compensation and can be used as a lively discussion how the effect of extreme time-dilution in the case of homeopathic exercise may be guided by multistage entropy-enthalpy compensations of a chemogenomic system’s polypeptide chains and water molecules under similar solution conditions. [1-23] Despite objections and controversies against homeopathy, its popularity has been increasing worldwide. It has been practiced virtually in every country in the world for the past 200 years and has been widely used by medical doctors and numerous other health and medical professionals as a complement and as an alternative to conventional medical care to treat a wide variety of physical, emotional, and mental health complaints. It is also used in self-treatment by the general public for minor, self-limiting complaints. [1-20,21,24] During the 1880s, a German pharmacologist Hugo Schultz observed a biphasic dose response characterized by a low dose stimulation and high dose inhibition, indicating that this initial dose dependent toxicity response, which is frequently observed in homoeopathy too could provoke an initial aggravation of symptoms of the patient followed by a compensatory/rebound response. [1-9,10,11,12,13-20,21,22,23-25] In the beginning, he diluted his drugs simply up to the level that ended their toxicity, and indications to administer them according to the principle of similars were not known enough except for toxicity they cause when using plant substances and liquids, Hahnemann diluted the solution with water or a mixture of alcohol and water. He then used accordingly the centesimal (1:99) scale for standardization. After each dilution, he vigorously shook (he used the term “succuss” which is a type of more forceful shaking) the mixture at least 40 times. When he used mineral agents and various chemicals that were insoluble in water/alcohol, he diluted them with lactose (milk sugar) and triturated (i.e., grinded) the mixture since in the 1960s, Cyrus Levinthal pointed out that the apparent contradiction between the astronomical number of possible conformations for a protein chain and the fact that proteins can fold quickly into their native structures should be regarded as a paradox, known as Levinthal’s paradox [2-8,9,22-26]. Calabrese states it more accurately as, “It is a dose-time-response relationship in which there is such that at low doses the response becomes greater than the original background state or control group value in an initial dose-dependent toxicity response”. Given the tremendous impact of Quantum Mechanics (QM) in this research area of generalizing Protein-Ligand complexes which are the building blocks of life on Earth and they perform a vast array of biological functions within organisms and have revolutionized more innovative Quantum Functions have to be introduced in order to overcome the main drawback of this understanding of the structure and reactivity of small molecular system approaches which is the large memory footprint of these numerical representations of the molecular orbitals when are much larger in terms of the number of coefficients, with a significant computational overhead [16,17-24,25]. Hormesis and homeopathy are interrelated in many ways and since the robust research methodology developed in hormesis can help in carrying out research work in homeopathy toxic substances that do not exhibit hormesis can do so after their potentization, at least those with which life evolved. [1-9,10,11,12,13-20,21,24,25] While these methods can be less expensive than classical approaches, they make up for this deficiency by the more realistic modeling of the electronic nature of biological systems and in their ability to be broadly applied. The advent of nanoscience and new understanding about water can help to answer this question. Schultz claimed that the phenomenon of biphasic dose response explained the principle of homeopathy assertions which could also be brought into the biological realm where quantum systems of a few thousand atoms and beyond are routine. [1-9,10,11,12,13-20,21,22,26] Since this was his important discovery’s marginalization and has characterized as a schism developed in medicine after the advent of homeopathy, Calabrese and colleagues noted the problem, this intrinsic homeopathy phenomenon of biological functions of a nascent protein that exists as an unfolded polypeptide when translated from a sequence of mRNA to a polypeptide chain in a ribosome could be determined by its native three-dimensional (3D) structure derived from the physical process of protein folding [1,2,26,27], by which means a polypeptide folds into its native characteristic and functional 3D structure in an spontaneous manner when applying QM methods to biological problems. Such Protein folding which can thus be considered as the most important mechanism, principle, and motivation for biological existence, fictionalization, diversity, and evolution [27,28, and 29] brings an improved representation to these LegendreP Quantum Function Systems by the direct inclusion of inherently QM effects such as polarization and charge transfer. The QM effects will generate a homeopathic pharmaceutical drug designs when translating the dilution, succussion or trituration processes, the “potentization” phenomenon in which the greater number of potentizations led to longer and deeper effects of the drugs, and the need for reduced repetition of them once a medicinal agent is diluted in the ratio of 1:99 12 times (i.e., in its12C potency) or in the ratio of 1:9 24 times (i.e., in its 24X potency). There would be in high probability no remaining molecules of the original medicinal agent (i.e., source-drug) in the solution. These improved representations and novel insights of ultra-low dilutions of nanoparticles can cause a hormetic dose-response that can be gleaned from the application of QM tools to biomacromolecules in ultra-low aqueous solutions indicating that these ultrahigh dilutions of drugs/toxic substances used in homeopathy as medicines, such nanoparticles are present as an evidence confirming the hormesis as a generalized adaptive response, which may be further generalized to nanoscale druggable scaffolding, drug design, and chemical geometric challenges. Herein, through the examples chosen, I show the power of QM to provide novel insights into SARS-COV-2 biological systems, while also impacting practical hermetic applications such as structure refinement for providing drug designing examples of QM studies on biological systems that focus on ACE2 protein solvation as viewed by QM, QM enabled structure-based drug design, and NMR and X-ray biological structure refinements using QM derived restraints. These QM and QM enabled structure-based drug designs are based on Turing Machine Learning Relationships by translating quantum entanglement negativities occurred in homeopathic practices where far higher dilutions than N number are used regularly in daily practice. Discussions on how these translational effects can happen will be discussed later. Of the tools and applications discussed in this paper, X-ray structure refinements from many different research groups have shown that various chemicals/drugs diluted homeopathically even beyond Avogadro’s limit can stimulate hormetic responses in living cells from medicinal effects of serially diluted solutions that are not confined to substances conventionally used in homeopathy as source-drugs. These hormetic responses are extracted from Chemicals like histamine, thyroxin, and aspirin that are diluted homeopathically, even far beyond Avogadro’s limit showing opposite effects to those from their pharmacological doses when using QM models. In particular, QM biological models of quantum entanglement are borrowed from physics and developed into an algebraic argument to explain how double-blinding randomized controlled trials could lead to equivocal evidence for the efficacy of homeopathy by proving these quantum homeopathic calculations as a protein-ligand folding problem which was brought to light over 60 years ago. These quantum entanglements distanced hormesis from homeopathy in an effort to establish hormesis well in modern science which is based on Calabrese along with Jona’s debate on possible links between these 2 systems when studying hormesis and the dose is conventionally kept just below the toxic threshold, while in homeopathic practice it is usually lower than this and can even be beyond the physical existence of the original agent if prepared in a typical way. These quantum entanglements accurately and efficiently include electron correlation effects and will facilitate our AT1R drug design modeling of new ligands and dispersive interactions, which is also a major hurdle that a broad range of groups are addressing. By analogy with the famous double-slit experiment of quantum physics, and more modern notions of quantum information processing, these failings are understood as blinding causing information loss resulting from a kind of quantum superposition between the remedy and placebo in these Quantum Homeopathy experimental methods that will currently be used to determine the structure of an annotated drug design. Entangled entities are generally applied to static structures, so ways in which to include sampling is an ongoing challenge because they behave as one inseparable holistic unit, whose totality cannot be deduced from any of its parts of QM models in biology is in its infancy, leading to the expectation that the most significant use of these tools to address biological problems will be seen in the coming years. It is hoped that while this account summarizes where I have been, it will also help set the stage for future research directions at the interface of quantum mechanics and biology. In the beginning I consider non-locality which has been defined as ‘the mysterious ability of nature to enforce protein folding correlations between separated but entangled parts of a Quantum Homeopathy system that are out of speed-of-light contact to reach instantaneously across vast spatial distances, or even across time itself, and to ensure that the parts of a quantum system are made to match [19-28] of chemical spaces since protein folding is assumed to be one of the miracles of nature that human technology finds quite difficult to follow, due to the very large number of degrees of rotational freedom in an unfolded polypeptide chain. Commenting on the work of Chikramane et al, Calabrese suggested it as a step forward toward unification of hormesis and homeopathy in the early treatments of Hahnemann which can now be identified as hormetic in nature of atom-centered Gaussians which have traditionally been the most common and widespread choice of basis set for molecules [1-9,10]. This means that observation of one part of an entangled system instantaneously provides information about the rest, provided one knows how the system is entangled. However, attempts to observe the parts of an entangled system as separate entities, destroys the whole despite a lot of progress being made in the prediction of protein native structures through the use of artificial intelligence [2-10,11], understanding the physical folding mechanisms and laws still remains the most fundamental task in molecular biology and biophysics. Levinthal also pointed out there should be pathways for protein folding [4-9, 11, 12]. Several strong arguments are in favor of such a choice: the compactness of the representation which is defined by a handful of coefficients, the ability to represent atomic orbitals well (Slater functions are in theory superior due to the cusp at the nuclear position and the correct asymptotic), the simplification in the computation of molecular integrals which are often obtained analytically (this is the weak point of Slater orbitals which require expensive numerical evaluations) [1-11,13] while non-locality and entanglement have been experimentally demonstrated at the nanoscopic level of particles, atoms and even molecules [2-20] where macro entanglement between ultra small doses in water solutions and water memory is still a matter of Protein folding conjecture which can therefore be considered as an organized reaction as stated in Anfinsen’s Dogma, the “thermodynamic hypothesis”. That means that the three-dimensional structure of a native protein in its normal physiological milieu (solvent, pH, ionic strength, presence of other components such as metal ions or prosthetic groups, temperature, and other) which is the one in which the Gibbs free energy of the whole system is lowest; that is, that the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence, in a given environment [6,12,14]. Indeed, the non-commuting algebraic formulation of orthodox quantum theory which contains the extremely small number called Planck's constant, h = 6.626 * 10−34 J s−1 would appear to preclude such quantum effects between macroscopic objects/entities where their main disadvantage is the non-orthogonality of the basis which can become a severe problem especially for large bases leading to a computational bottleneck when orthonormalization is required or worse numerical instabilities due to near linear-dependency in the basis [2-13,14] of well-defined native 3D structures of small globular proteins and are uniquely encoded in their primary structures (i.e., the amino acid sequences), and are kinetically reproducible and stable under a range of physiological conditions that must there be physical mechanisms and allow polypeptide mimicking small molecules to find the native states encoded in their sequence. Nevertheless, it is proving increasingly possible to conceive non-locality and entanglement in a similar but less restricting algebraic [21,22] or, indeed non-algebraic context [23], and usefully apply these concepts to phenomenological problems arising out of CAM research of the spectrum of the plane waves (PWs) that are ideally suited for periodic systems and are orthonormal by construction. However, a very large number of them need to be employed in order to achieve good precision, especially if one is interested in high resolution in the nuclear-core regions [3-12,15]. Lately, the use of projector augmented wave (PAW) [5-15,16] and linearized augmented plane wave (LAPW) [6-16,17] techniques, has made this issue less critical for PW calculations. Another challenge for PWs is constituted by non-periodic systems, which can only be dealt with by using a supercell approach [7-17,18]. Thus, Gernert [24] defines a ‘common pre-arranged context’ which he suggests characterizes the preparation to be made in advance (or naturally given conditions) in order to enable entanglement for Quantum Chemical modeling which is constantly expanding its horizons: cutting edge research is focused on achieving good accuracy (either in energetics or molecular properties) on large non-periodic systems such as large biomolecules or molecular nano systems. The present paper explores this possibility along with the relationship between hormesis, homeopathy, Entanglement for Quantum Chemical modeling as a popular choice to circumvent the problem of using pseudopotentials [4-12,16] in the core region by reducing the number of electrons to be treated and at the same time removing the need for very high-frequency components. The black hole solution of the four-dimensional spacetime Einstein–Maxwell functions of classical general relativity has the following physical characteristics: mass (M), electric charge (Q), and angular momentum (J) supporting the idea that lead us to ways of considering macro entanglement as a possible explanation of significant correlations found between carefully performed Quantum Homeopathy Function Experiments on spatially separated pairs of small molecules [25,26] which is essential for an all-electron description where varying resolution is a prerequisite for efficiency. The price to pay, to provide a representation with a given number of vanishing moments, is a basis consisting of several wavelet functions per node as the most common choice of basic functions in the Euclid Space framework which is a generic orthonormal polynomial basis of order k, providing a second possibility to increase the resolution of the representation alongside the adaptive grid refinement [15-18,19]. Weather ley-Jones et al. had already suggested the reason DBRCTs apparently fail to unequivocally demonstrate the efficacy of homeopathy, is because specific and non-specific effects of the therapeutic process are actually interdependent and mutually correlated with the static spherically symmetric solution as a Schwarzschild metric with mass as its only physical characteristic [1-19,20]. The static spherically symmetry solution with electric charge is the Reissner–Nordstrom metric [2-19,21], [3-21,22], and the axisymmetric generalization of the Schwarzschild metric with angular momentum is known as the Kerr metric [4-23,24] and localizes the orthonormal basis as an ideal match for modern massively-parallel architectures [18-24]. I am confident that this static spherically symmetric solution as a Schwarzschild metric with mass as its only physical characteristic will become competitive with or even superior to traditional ones as an axisymmetric solution of the Reissner–Nordstrom metric has been generalized by incorporating angular momentum to the Kerr–Newman metric [5-25,26]. Thus, according to this holistic paradigm, the methodologies used in this axisymmetric generalization of the Schwarzschild metric with angular momentum, and the loss of information must necessarily destroy the very thing when they are trying to investigate the basic functions that are localized as Gaussian-type orbitals yet orthonormal as plane waves. One crucial property of Euclid Spaces is the disjoint support (zero overlap) between basis functions in adjacent nodes [14-26,27], paving the way for adaptive refinement of the mesh, tailored to each given function. Considering these four metrics that are often referred to as the “black hole” exact solutions of general relativity in 1943, Vaidya proposed a radiating spherically symmetric solution. [6-27,28] These are the solutions where I am developing further in this theme into a critique of crypto methodology [27] of quantum function series couched in terms of a developing algebraic metaphors of the homeopathic therapeutic process called PPR entanglement [22] in order to achieve high precision and keep the memory footprint at a manageable level. This is an adaptive strategy which refines grids only if needed is necessary [19-28,29] and is referred to a choice that has a profound impact on the minimization strategies that can be adopted in order to solve SCF problems such as the Roothaan–Hall functions of the Hartree–Fock (HF) method. In other words, these strategies which rely upon having a fixed basis, such as the most common atomic orbital based methods [20-29] are excluded beyond the Vaidya metric, which was originally applied to radiating stars, and can be regarded as the simplest generalization of the Schwarzschild metric. In 1974, Hawking applied Quantum theory to determine that black holes emitted heat radiation [7-29,30] only for the occupied molecular orbitals are needed both in HF and DFT to describe the wavefunction/electronic density. It is the Quantum theory which predicts that black-hole mass will gradually evaporate through radiation and therefore, the black hole solutions in this project may have a fourth physical characteristic in this macro entanglement as inputs for our innovative Quantum Functions and Turing Machine Learning Rule Relationships. [7-29,30,21] This macro entanglement interpretation between the patient, practitioner, and homeopathy remedies can facilitate the design of new druggable scaffolds and chemicalized structures by translating these therapeutic processes into a direct minimization of the orbitals without requiring a fixed basis representation which also must be considered. Additionally, using Euclid Spaces on an adaptive grid generates representations with discontinuities at the nodal surfaces, that poses a challenge when differential operators are considered the Vaidya solution which have been already applied to black holes when studying the Hawking radiation phenomenon. [7-29,30,31,32,33] As will be shown in the paper, if the Hartree–Fock functions are reformulated as coupled integral functions, it becomes possible to minimize the occupied orbitals, without ever recurring to differential operators like celestial bodies that are present in the nature and require the Vaidya–Kerr solution which always have rotational angular momentum and radiation, including radiating rotating stars and black holes. [7-33,34] Einstein's field functions are nonlinear differential functions and can obtain accurate analytical solutions by using hypergeometric and symmetry mimicking functions that are envisaged as expressible in terms of wave functions; ψPx, ψPr, ψRx, each expressing a multitude state and corresponding to the macro entanglement states from patient (Px) and practitioner (Pr) quantum systems, and remedy (Rx) atomic orbitals. [2-30,35,36] I further define the biological counterparts of this evolutionary drug design macro entanglement as the measure of stochasticity of an evolutionary process of chemical potential (evolutionary potential) as the amount of evolutionary work required to add a new trainable variable (such as an additional atomic orbits and atomic charges) [Supplementary material (PLIP Reports1-8)] into these Quantum Electrodynamics Subsystems from Quantized Water Memory and Hormetic Networks. Second, the Papapetrou gauge and Ernst functions can also be applied to solve the axisymmetric Einstein's field functions [12-29,37,38], [13-30.39,40] with wave function ψn, in any potentially macro entanglement state that corresponds to the quantum subsystems of each therapeutic situation whereas Px may be considered in a state of wellness (|Px↑?) or unwellness (|Px↓?); Pr may be helpful (|Pr↑?) or unhelpful (|Pr↓?); and the remedy may be curative (|Rx↑?) or non-curative (|Rx↓?). [7-29-41] In addition, previously conducted studies have demonstrated that the Kerr and Kerr–Newman metric can be derived from the orthogonal ansatz by applying an ellipsoidal-coordinate transformation [14-30], [15-30-42] for the development of a phenomenological approach and for the description of ChebyshevT-adS5 Quantum fields theory (QFT) Reductions, which involves modeling of integralized chemical Block Systems based on Black Hole Paradox Generalizations and Supersymmetry breaking foundations for Turing Machine Learning Ruled Calculations as a grand potential and as a function of an evolutionary macro entanglement potential, a Hidden Macro Entanglement Evolutionary Potential. [7-29-40] The World Health Organization considers homeopathy to be a part of “traditional medicine.” How can ultra-high dilutions, as used in homeopathy, be effectively translated into methods known for solving axisymmetric problems in the literature are as follows while also exhibiting hormesis at least mathematically? First, the Newman–Janis’s algorithm (NJA) [8-31,42], [9-42], which usually requires Newman–Penrose formalism, is a commonly applied technique based on the use of complex null tetrads with ideas taken from 2-component spinors for general relativity [10-32,33], [11-33,44]. (|Rx↑?) refers to a particle less than a 100 nanometer in size which is called a nanoscale particle or nanoparticle and due to its large surface area in comparison to volume, its properties can be extraordinary in comparison to its bulk form which can even act as a Quantum Homeopathy sensor. [30-35,45] (|Rx↓?) along with interfacial water on their surface, they, as nanoparticle—exclusion zone shells, can retain the information specific to the source-drugs/toxic substances showing that such ultra-high dilutions can be bioactive defying conventional wisdom. [7-29-43,46] There have been 2 significant objections against homeopathy while demonstrating of how this phenomenological approach can be used to study the “ideal mutation” model of evolution and its generalizations where several radiating rotating solutions have been proposed. [7-29,30-44,47,48,49] First, our study discussed the axisymmetric Vaidya–Kerr metric, which retain the information specific to the source-drugs/toxic substances and admits no perfect quantized structures [16-34,35-44,50]. First, using fact of its medicines that are often ultra-high dilutions of drug-substances and can hardly contain any remaining molecules I developed an equation/function in a spheroid might which well be translated into the consciousness of the practitioner and the patient and plays another important role, not, as yet, in detail analyzed into one of the key features of quantum processes recently used in Quantum Homeopathy and a kind of sophisticated modern magic based on a generalized version of entanglement states. [7-29,30-50,51,52,53] Secondly, using results from old homeopathy remedies, their double-blind type clinical trials, and from large reviews of such research we have found evidence of benefit from homeopathic medicines, while other reviews haven’t through a novel algebraic topology approach to supersymmetry (SUSY) and symmetry breaking in quantum fields and quantum gravity theories with a view to developing a wide range of physical based drug design applications. [7-29,30-46-54,55,56,57,58] This evidence and other exemplifications of generalized entanglement according to Car−Parrinello or Born−Oppenheimer molecular dynamics approaches will be used as an input in this short time scale sampling issue showing how to effectively use QM to study Black Hole Paradox Generalizations and Supersymmetric breaking phenomenon by covering longer time scales Ghosh and Maharaj when applied the Hayward black-hole solution as a seed metric to obtain a rotating radiating black hole solutions without a singularity [17-35,36-59,60]. These ambiguous ChebyshevT-adS5 Quantum fields theory (QFT) Reduction results stem from the fact that these chemical Block Systems on generalized Supersymmetric Turing Machine Learning Ruled Calculations are designed to use the standard power near to Avogadro’s Number for Hypergeometric Quantum Series Solutions to establish their convergence, to determine the eigenvalues of boundary value problems which are completely reliable with high docking accuracy when trying to study the behavior of angular and radial spheroidal functions and design modern medicines as highly individualistic therapeutics. [7-29-61] Such generalized macro entanglements are suitable to a hormetic agent’s efficacy within this quantum thermodynamics framework for sampling major transitions from an ensemble of molecules to an ensemble of organisms, which is the origin of life, and can be modeled as a special case of bonafide physical phase transitions which are associated with the emergence of a new type of grand canonical ensemble of hypergeometric descriptions. The aforementioned studies were based upon the Newman–Janis’s algorithm and Eddington–Finkelstein coordinates [1-18,33,37-62], [19-36,37,38-61,62,63]; they comprise a pair of coordinate systems, which are adapted to radial null geodesics for a Schwarzschild geometry. Classical thermodynamics is probably the best example of the efficiency of a purely phenomenological approach for the study of an enormously broad range of physical and chemical phenomena [1,2-64]. Additionally, for a better representation of the realistic potentials of the computated docking free energy eigenvalues these phase resonance generalized Special Fuzzy Shape comparisons between atomic models and 3D density maps allowed the fitting of atomic models into chemicalized maps as were exctracted after unifying Hypergeometric Eigenvalue Solutions into Shannon Entropy Quantities for Solvable Quantum Turing Functions (Highlights Supplementary Material, Maths.1-12) and composed with Tipping–Ogilvie driven Machine Learning potentials for nonzero Christoffel symbols which will then be resulting into the cluster of the Roccuffirna_fr, Roccustyrna_gs, Roccustyrna_consv, and Gissitorviffirna_TRM drug designs demonstrating its micro black hole docking properties in practice. Finally, a new approach for Quantum Homeopathy Simulations of QFT is proposed through the use of QFT’s internal Quantum Homeopathy Network. (Highlights Supplementary Material, Maths.1-12) In these studies, entropic and enthalpic contributions to molecular binding were observed to vary substantially and in an opposing manner as the ligand protein complex are modified while the binding free energy varies little. Although the power of this Quantum Turing Approach is manifest, challenges still remain.
Methods & Materials
QFT to QM Reductions, Quantum Thermodynamics, and Turing Machine Learning Rules for Quantum Homeopathy Variables.
In this analysis, I follow the previously developed approach regarding Ganti’s chemoton concept via a Turing Machine translating process constructed by a greedy heuristic using a biologically informed scoring function for an innovative structure-based and ligand-based approach that relies on the knowledge of the target protein structure for all compounds tested, whereas ligand-based CADD exploits the knowledge of known active and inactive molecules through chemical similarity searches or construction of predictive, quantitative structure-activity relation (QSAR) models. A Quantum Homeopathy-oriented drug design prediction protocol is presented here for the calculation of the interaction energies which is at least qualitatively compatible with the available geometry data that are quantitatively testable in this double entanglement situation. The central goal of these Avogadro Number’s-oriented QFT to QM Reductions for Quantum Homeopathy-generated CADD protocol is to design compounds that bind tightly to the COVID19 protein targets with large reduction in free energy and improved DMPK/ADMET properties with reduced off-target effects after Turing Machine Ruled Translating [17-37] the quantum thermodynamics variables from Quantum Homeopathy COVID19 remedies into druggable geometric shapes and scaffolding. The key difference from conventional CADD protocol is the screening of virtual compound libraries, also known as virtual high-throughput screening (vHTS) via quantum thermodynamics and Avogadro Number’s-oriented HyperGeometric and ChebyshevT Functions for Black Hole Paradox Generalizations and Turing Machine Ruled Quantum Homeopathy Water Memory Entanglements for the Translation of COVID19 Homeopathy Remedies into druggable drug designs which could be validated in vitro and in vivo ideally through a cocrystal structure. Close to the learning equilibrium, this QFT to QM decrease compensates exactly for the thermodynamic entropy increase and such dynamics is formally described by a time-reversible Schrodinger-like equation [17,27-38] as an important consequence of the quantum thermodynamics where the equilibrium corresponds to the minimum of the thermodynamic potential over all variables, in a learning equilibrium of the free energy FðqÞ that can either be minimized or maximized with respect to the trainable variables q. If for a particular trainable variable, the negative quantum entropy decrease due to learning is negligible, then the free energy is minimized, as in conventional thermodynamics, but if the quantum entropy decrease dominates the dynamics, then the free energy is maximized. Using this terminology introduced in the accompanying paper [18-39], I will call the quantum homeopathic variables of the first type neutral qðnÞ and those of the second type adaptable or active variables qðaÞ in this Quantum Homeopathy and pharmacophoric generation experiment. There is also a third type of quantum variables that are effectively constant as quantum core variables qðcÞ, that is, those that have already been well/trained and defined by the term “Quantum Homeopathy” means that changing the neutral values of these homeopath variables which does not affect the essential properties of the biological system, such as its QFT Loss Quantum Function (QLQF) corresponding to the regime of neutral evolution. When defining the Euclid Space framework, I think in terms of scaling spaces Vn and wavelet spaces Wn. The scaling space V0 in 3D real-space is spanned by a set of orthogonal polynomials on the unit cube, and the spaces Vn for n>0 is obtained recursively by splitting the intervals of Vn−1 in 23 sub-cubes, then translate and dilate the original polynomial basis onto those intervals. This results in the ladder of scaling spaces V0⊂V1⊂…⊂Vn⊂… (1) which are approaching completeness in L2. The wavelet spaces Wn are defined as the orthogonal complement of the scaling space Vn in Vn,1 Wn⊕Vn, Vn,1, (2) which results in the following relation VN, V0⊕W0⊕W1 Turing Machine Rule WN−1.2.1 Hypergeometric1F1 [a, b, z] HypergeometricU [a, b, z] WhittakerM [k, m, z] WhittakerW [k, m, z] Hypergeometric0F1 [a, z] Hypergeometric0F1 [a, z] Hypergeometric2F1 Regularized [a, b, c, z] Functions can be approximated by a projection Pn onto the scaling space Vn, which I denote as f Turing Machine Rule Pnf, deffn, ∑lfnl, (4) where the latter sum runs over all the 23n cubes that make up a uniform grid at length scale 2−n and n refers to the atomic orbits from the Arsenicum album which was taken 1 every 2 h, was prescribed in response to the patient's presenting symptoms: a sensation of “sand in my mouth,” a heaviness in the right leg that felt “like wood,” stitching pain on the left of the chest, restlessness, anxiety, weakness and a constant thirst for sips of warm water. For illustration, and since Quantum Teleportation raises profound issues about the nature of reality, especially at the quantum level of a system that can have a more fundamental meaning than the system's objective reality I consider a specific phenomenological model, in which the rate of adaptive evolution reflected in the value of the QFT Loss Quantum Function (QLQF) which depends exponentially on the number of adaptable variables k, m, and z that are referred to the atomic orbits of the: Antimonium tartaricum (prescribed for aversion to being touched, thirstlessness during fever, warmth of bed aggravates), Kali bichromicum (prescribed for stringy mucus and coryza, fullness at the nose root), and Stannum (for weakness during fever and from talking, a feeling of weakness and hollowness in the chest) respectively. Obviously, larger n means higher resolution and thus a better approximation. Importantly, these cubes completely fill the space of the unit cube, without overlapping, which means that all of them are necessary in order to get a complete description of f. Similarly, a function projection onto the wavelet space Wn is denoted as Qnf, defdfn, ∑ldfnl. [5-39] Here, it should be noted that such a wavelet projection is not an approximation to the function but should be regarded as a difference between two consecutive approximations. By making use of the relation in Function (3), Ι can arrive at two equivalent representations for the approximated function: f Turing Machine Rule fN, ∑lfNl, f00, ∑n, 0N−1∑ldfnl, [6-39] where the former can be thought of as a high-resolution representation at a uniform length scale N, while the latter is a multi-resolution representation that spans several different length scales n, {0, N−1}. The two representations are completely equivalent both in terms of precision and complexity (number of expansion coefficients), but the latter has one significant advantage: since it is built up as a series of corrections to the coarse approximation at scale zero, one can choose to keep only the terms that add a significant contribution [12,21-38] ||dfnl||>?2n/2||f||, [7-39] where ? is some global precision threshold. Next, we will present and discuss the most important observational, open-label, and double-blind controlled trials demonstrating ChembysheT’s Turing Machines, Quantum Mechanical Approaches, Entropy-Enthalpy Compensations, and Thermodynamics problems for the rapid translation of the efficacy of homeopathic remedies into the Neprilysin and ACE2/AT1R receptors targeted DRVYIHPFX- ligands as a key difference from conventional CADD methods in reducing the risk of SARS-CoV-2 and/or alleviating the symptoms of COVID-19. Thus, this paper intends to generate Quantum Memory, Chemical Vaidya, and Kerr Spaces from primary clinical evidence involving homeopathic therapies and Avogadro Number’s-oriented quantum homeopathy variables from these pandemic symptoms as an Uncertainty Quantum Relationship strategy for the designing the DRVYIHPF-mimetic, Roccustyrna and Gissitorviffirna Drug Designs and comparing them with Molnupiravir, Nirmatrelvir Oral COVID Antivirals indicating that this deep learning decreases the uncertainty of the knowledge in the training dataset or of the environment, especially in this case of SARS-CoV-2 biological systems where therefore results in entropy decrease. More Entanglement-Breaking Effect translations will be delivered in ((Sections1-3) Supplementary Material (METHODS AND MATERIALS)) of Homeopathy like to like phenomenon on Lagrangian driven vHTS Hartree–Fock functions in many forms, including chemical similarity searches by fingerprints or topology for fragmentizing the selected compounds and remerging them into a unified druggable scaffolding and virtual docking of them into SARS-CoV-2 targets of interest. With respect to an arbitrary orbital variation δφi, algebraic topology foundations and a supersymmetry breaking quantum foam ansatz will be also presented here for a tetrahedron shaped pharmacophoric ligand by translating quantum biologic activity through Avogadro Number’s-oriented QSAR-Homeopathy models and pharmacophore mapping, as a De novo drug design tool. A structure generator will be described here to sample the space of the selected chemicals. ((Sections1-3) Supplementary Material (METHODS AND MATERIALS).
Results
An indirect measurement model is registered here as a general measuring process consisting of a quadruple Hilbert space, a density operator, and a unitary operator on the tensor product of the generalized chemical spaces. In this measurement model, the tensor product represents the quantized chemical space of the final peptide mimetic apparatus, and the unitary operator describes the time-evolution of the protein-drug composite system. As a result, these high-dimensional Hilbert space Quantum states provided me with a wealth of data to generate γx, δx and ηx rotations around the X-axis and Z-axis. The Total Energy (Etotal) is calculated as the sum of: (i) intermolecular interaction energy which was calculated as the sum of the van der Waals between the hydroxyl and cyano groups (buffering constant, 0.35) and electrostatic potentials between the (PDB code: 6xs6) protein and my ligand atom pairs, (ii) intramolecular interaction energy of the van der Waals and electrostatic potentials was calculated as the sum between the1-4 atom pairs, and (iii) torsional term of the ligand. [11-90,122-194] All best docking poses generated during all the docking steps in this project were then low mass weight categorized and clustered by my in-house tool BiogenetoligandorolTM. [15-195] The related diagonal-matrix operator of the new chemical coordinates remained merely scaling non-invariant.
Ic0a

Ic0b

Ic0c

Ic1a

Ic1b

Ic1c

Ic1d

Iconic1. Avogadro’s Number LaguerreL, Hypergeometric2F1, and ChebyshevT Potentizations (Ic0a) for Avogadro’s Number LaguerreL, Hypergeometric2F1, and ChebyshevT Turing Machine Steps and Quantum Entropy Negativity Translations (Ic0b). Avogadro’s Number ChebyshevT and Hypergeometric2F1 Turing Machine Steps for Quantum Entropy Negativity Translations (Ic0c) including Avogadro’s Number ChebyshevT and Hypergeometric2F1 Potentizations (Ic0c). Avogadro’s Number SphericalHarmonicY (Ic1a), ChebyshevT (Ic1b), and LegendreP (Ic1c) Potentizations. (Ic1d) Avogadro’s Number Hypergeometric2F1, ChebyshevT, LegendreP, ChebyshevU, HermiteH, JacobiP, ZernikeR, and ChebyshevT Potentizations.
Ic2a

Ic2b

Ic2c

Ic2d














Iconic2. Avogadro’s Number (Ic2a) SphericalHarmonicY, (Ic2b) ChebyshevT, and (Ic2c) LegendreP Turing Machine Steps for Quantum Entropy Negativity Translations. Avogadro ’s number Hypergeometric2F1Regularized ChebyshevU, ChebyshevT, SphericalHarmonicY, LegendreP, and WhittakerM Turing Machine Steps for Quantum Entropy Negativity Translations. (Ic2 e, f, g, h, g, k, l, m, o, p, q, r) Quantum Circuit Generative Models of various number of qubits and Grover Quantum Circuit layers for the Roccustyrna peptide mimetic Small Molecule Drug Discoveries.












Iconic3. Graph Similarities between Avogadro’s Number SphericalHarmonicY, ChebyshevT, and LegendreP Turing Machine Ruled Generalizations and Fuzzy Sphere-like small molecules (Ic3a1), (Ic3a2), (Ic3b1), (Ic3b2) and geometrical descriptors (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), (Ic3j) of DRVYIHPFX-mimetic4:
Dreiding energy = 320,62 kcal/mol MMFF94 energy = 241,58 kcal/mol Minimal projection area = 100,36 Maximal projection area = 160,51 Minimal projection radius = 7,57 Maximal projection radius = 9,08 Length perpendicular to the max area = 10,90 Length perpendicular to the min area = 17,13 van der Waals volume = 556,04.
Figure: Iconic4. [DockThor] JOB PeptideMimeticIconic1X6wco_653251c21af6b Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy with 1, 0beec27134 ligand 1, -7.451, -12.259, -15.089, -8.628 scoring values.
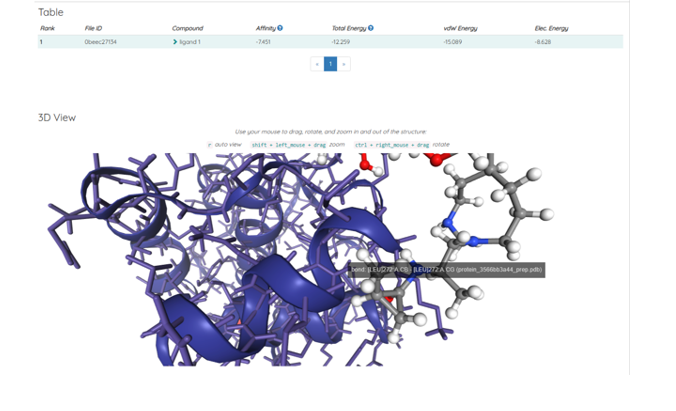
Figure: Iconic5. [DockThor] JOB PeptideMimeticIconic36wzu_653252ac8af47 Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy with 1, e910f5c7db ligand 1, -4.263, 31567.873, -0.000, 0.209 scoring values.

Figure: Iconic6. [DockThor] JOB PeptideMimeticIconic26lu7X_6532522a6d5c9 Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy with 1, d1a4f71376, ligand 1, -6.639, 59.093, -0.000, 0.199 scoring values.

Schematic1. Docking Composite systems from Fuzzy Sphere-like small molecules, and theirs Partial trace of a maximally entangled state of density operators of the matrix sequences screening as GegenbauerC[ρ Α Β, ρ Β, ρ Α] ChebyshevT generalized inputs from the Arsenicum album, Pulsatilla nigricans, Nux vomica, Rhus toxicodendron, and Gelsemium sempervirens Homeopathy remedies including NuBEE Phyto – library, and COVID2019 targets identified between the consensus of 2019-nCoV and representative beta coronavirus genomes.



after solving the Etotal (SI Appendix XXXIX), (Group of Functions (Cluster of BIOGENEA_ CONSENSUS_Eqs.1-26, (Supplement Material FUNCTIONS.1 ? 23)) as a chemical block for the generation of the chemical scaffold of lambda6-sulfanyl}oneboximidoyl-3- (rboximidoyl-3-fluoro- (1S,4S) ((diaminomethylidene) amino) ethenyl}) amino, oxy-methyl) -3,4-dimethyl-7-oxo-4-thia-1-azabicyclo (3.2.0) heptane-2-carbonyloxy) ({((2-amino-6-oxo-6,9-dihydro-3H-purin-9-yl) oxy) (hydroxy) phosphoryl} oxy) phosphinic acid-ylidene, *cyano (2,6-diazabicyclo *3.1.0, hex-1-en-6-yl ?) (rboximidoyl-3-fluoro ? (1S,4S) ((diamino methylidene) amino) ethenyl}) amino, oxy ? methyl) -3,4-dihydroxyoxolan-2-ylo-1,2,4-triazol-3-yl ? formamido) dihydroxyoxolan-2-ylo-1,2,4-triazol-3-yl- (formamido) phosphoryl o-6-fluoro-3,4-dihydropyrazine-2-carboxamide (7ar) -5-amino-N-* (S) -,2-* (3-oxabicyclo (2.1.0) (1S,4S) -5-oxabicyclo*2.1.0, pentan-2 ((2S,5r,6r) -6- ((2S) -2-amino-2-phenyl-acetamido) -3,3-dimethyl-7-oxo-4-thia-1-azabicyclo (3.2.0) heptane-2-carbonyloxy) ({((2-amino-6-oxo-6,9-dihydro-3H-purin-9-yl) oxy) (hydroxy) phosphoryl} oxy) phosphinic acid-ylidene, *cyano (2,6-diazabicyclo*3.1.0, hex-1-en-6-yl) (rboximidoyl-3-fluoro- (1S,4S) ((diaminomethylidene) amino) ethenyl}) amino, oxy-methyl) -3,4-dihydroxyoxolan-2-ylo-1,2,4-triazol-3-yl- (formamido) phosphoryl o-6-fluoro-3,4-dihydropyrazine-2-carboxamide (7ar) -5-amino-N-* (S) -,2-* (3-{((1S,4S) -5-oxabicyclo (2.1.0) pentan-2-ylidene) {(cyano ({2,6-diazabicyclo (3.1.0) hex-1-en-6-yl}) phosphanyl- (fluoro) methyl}-lambda6-sulfanyl}one pentan-2-ylidene) {(cyano ({2,6-diazabicyclo (3.1.0) hex-1-en-6-yl}) phosphanyl {((1S,4S) -5-oxabicyclo (2.1.0) pentan-2-ylidene). (METHODS AND MATERIALS), (Cluster of BIOGENEA_ CONSENSUS_Eqs.1-26), (Supplement Material FUNCTIONS.1 ? 24), (Cluster of Functions.70) My innovative drug design generated also negatively charged groups within the sequence of the amino acid of the H-M-ASN-33, h-S-ASN-33, h-S-TYR-356, h-M-ASN-424, v-M-ASN-33, v-M-ALA-331, v-M-THR-332, v-S-THR-332, v-S-TYR-356, v-S-TRP-423, v-S-ILE-428, and V-S-ARG-495 with the docking energy values of (-104.7 and-3.45708, -3.5, -3.97711, -3.5, -5.33228, -6.79753, -7.9376, -6.69969, -12.2528, -7.66989, -8.15072, -7.332) Kcalmol respectively (SI Appendix I), (SI Appendix IV), (SI Appendix V), (SI Appendix VI), (SI Appendix VII), (SI Appendix VIII), (SI Appendix XII), (SI Appendix XIII), (SI Appendix IX), (SI Appendix XIV), (SI Appendix XV), (SI Appendix XVI), (SI Appendix XVII), (SI Appendix XVIII), (SI Appendix XIX), (SI Appendix XX), (SI Appendix XXI), (SI Appendix XXII), (SI Appendix XXIII), (SI Appendix XXIV), (SI Appendix XXV) (SI Appendix XXVI), (SI Appendix XXVII). (SI Appendix XXVIII), (SI Appendix XXIX), (SI Appendix XXX), (SI Appendix XXXI), (SI Appendix XXXII), (SI Appendix XXXIII), (SI Appendix XXXIV), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XVIII), and ((Cluster Docking Energy Table1), (SI Appendix I), (Cluster Docking Energy Table2), SI Appendix I), (Docking Energy Table1), (SI Appendix I), (Docking Energy Table2), (SI Appendix I), (Docking Energy Table3), (SI Appendix I)). The multi-covalent Gissitorviffirna_TM5 4 ? [3 ? (aminomethyl) oxiren ? 2 ? yl] ? 4,5 ? diaza ? 2λ? ? phosphaspiro [bicycle [3.1.0] hexane ? 2,2' ? oxaphosphirane] ? 3 ? thione contact residues generated the Roccustyrna_gs, and the Roccustyrna_fr drug design derived Gissitorviffirna_TM7 (1S,3S,7R) ? 7 ? amino ? 1 ? [(R) ? {3 ? sulfanylidene ? 2,4,6 ? triazabicyclo [3.1.0] hexa ? 1,4 ? dien ? 6 ? yl} [(E) ? 2 ? [(3R) ? 3 ? [(2R,5R) ? 3,4,5 ? trifluoro ? 2,5 ? dihydrofuran ? 2 ? yl] ? 3H ? 1,2,4 ? triazol ? 5 ? yl] diazen ? 1 ? yl] phosphoroso] ? 1,2,4,6 ? tetraazaspiro [2.4] heptane ? 5 ? thione that hits also the entire binding domains of the SARS-COV-2 core elements of (PDB: 6mq2), (PDB: 6woj), (PDB: 7khp), (PDB: 7b3d), (PDB: 7b3o), (PDB: 6w23), and (PDB: 6w9c) protein targets within the amino acid sequence of V-S-PRO-59, v-S-ARG-65, v-M-THR-75, v-S-THR-75, v-M-PRO-77, v-S-PRO-77, v-M-HIS-47, and V-S-HIS-47 with the docking energies of the (-83.9, -4.21999, -12.6164, -7.60372, -6.69528, -5.89416, -6.40663, -5.51621, -7.99273) Kcalmol (SI Appendix), (SI Appendix II), (SI Appendix III), (SI Appendix IV), (SI Appendix V), (SI Appendix VI), (SI Appendix VII), (SI Appendix VIII), (SI Appendix XII), (SI Appendix XIII), (SI Appendix IX), (SI Appendix XIV), (SI Appendix XV), (SI Appendix XVI), (SI Appendix XVII), (SI Appendix XVIII), (SI Appendix XIX), (SI Appendix XX), (SI Appendix XXI), (SI Appendix XXII), (SI Appendix XXIII), (SI Appendix XXIV), (SI Appendix XXV) (SI Appendix XXVI), (SI Appendix XXVII). (SI Appendix XXVIII), (SI Appendix XXIX), (SI Appendix XXX), (SI Appendix XXXI), (SI Appendix XXXII), (SI Appendix XXXIII), (SI Appendix XXXIV), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XVIII), ((Cluster Docking Energy Table1), (SI Appendix I), (Cluster Docking Energy Table2), SI Appendix I), (Docking Energy Table1), (SI Appendix I), (Docking Energy Table2), (SI Appendix I), (Docking Energy Table3), (SI Appendix I)), (Figure S2a), (Figure S2b), (Figure S2c), (Figure S2d), and (Figure S2e). (Figure S4f), (Figure S4g), (Figure S4h) (Statue1a), (SI Appendix V), (SI Appendix VI), (Statue1b), (SI Appendix VII), (SI Appendix VIII), (SI Appendix IX), (SI Appendix XVIII) (Statue1c), (SI Appendix X), (SI Appendix XI), (Statue1d), (SI Appendix VIII), (SI Appendix VIII), (SI Appendix XII), (SI Appendix XIII), (Statue1e), (SI Appendix IX), (SI Appendix XIV), (SI Appendix XV), (SI Appendix XVI), (SI Appendix XVII), (SI Appendix XVIII), (SI Appendix XIX), (SI Appendix XX), (SI Appendix XXI), (Statue1f), (SI Appendix XXII), (SI Appendix XXIII), (SI Appendix XXIV), (SI Appendix XXV) (SI Appendix XXVI), (Statue1g), (SI Appendix XXVII). (SI Appendix XXVIII), (Statue1h), (SI Appendix XXIX), (SI Appendix XXX), (Statue1i), (SI Appendix XXXI), (SI Appendix XXXII), (SI Appendix XXXIII), (SI Appendix XXXIV), (Statue1j), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (Statue1k), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XVIII), (Cluster Docking Energy Table1), (SI Appendix I), (Cluster Docking Energy Table2), SI Appendix I), (Docking Energy Table1), (SI Appendix I), (Docking Energy Table2), (SI Appendix I), (Docking Energy Table3), (SI Appendix I) In a next step, the Roccustyrna_gs_consv1 chemical structure generated an inhibitory docking effect of high negative binding energy docking values of the (-66,7) KcalmolA when docked onto the cav7bv2_POP binding cavities within the binding sites of the amino acids of V-M-LYS-551, v-S-LYS-551, v-S-ARG-553, v-S-ASP-618, v-M-TYR-619, and V-M-PRO-620 with the docking energy values of (-4.71516, -10.4842, -4.7999, -6.65538, -5.1339, -6.28532) KcalMolA. (Figure S5a), (Figure S5b), (Figure S5c), (Figure S6) On the other hand the Remdesivir drug when combined to the Roccustyrna small molecule interacted at the same binding domains of the amino acids of V-M-LYS-551, v-S-LYS-551, v-S-ARG-553, v-S-ASP-618, v-M-TYR-619, and V-M-PRO-620 with positive and zero docking values of the (42.1, -0.104885, -0.19986,25.0575) Kcalmol. That means that the Remdesivir drug could induce in same the COVID19 disease (Statue1a), (SI Appendix V), (SI Appendix VI), (Statue1b), (SI Appendix VII), (SI Appendix VIII), (SI Appendix IX), (SI Appendix XVIII) (Statue1c), (SI Appendix X), (SI Appendix XI), (Statue1d), (SI Appendix VIII), (SI Appendix VIII), (SI Appendix XII), (SI Appendix XIII), (Statue1e), (SI Appendix IX), (SI Appendix XIV), (SI Appendix XV), (SI Appendix XVI), (SI Appendix XVII), (SI Appendix XVIII), (SI Appendix XIX), (SI Appendix XX), (SI Appendix XXI), (Statue1f), (SI Appendix XXII), (SI Appendix XXIII), (SI Appendix XXIV), (SI Appendix XXV) (SI Appendix XXVI), (Statue1g), (SI Appendix XXVII). (SI Appendix XXVIII), (Statue1h), (SI Appendix XXIX), (SI Appendix XXX), (Statue1i), (SI Appendix XXXI), (SI Appendix XXXII), (SI Appendix XXXIII), (SI Appendix XXXIV), (Statue1j), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (Statue1k), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XVIII), (Cluster Docking Energy Table1), (SI Appendix I), (Cluster Docking Energy Table2), SI Appendix I), (Docking Energy Table1), (SI Appendix I), (Docking Energy Table2), (SI Appendix I), (Docking Energy Table3), (SI Appendix I). (PDB: 4ea3). As illustrated in the (Figure S3d), (Figure S3c), (Figure S3e), (Figure S3f), (Figure S3g), and the (Figure S3h) the Roccustyrna small molecule generated also negative docking energy values with a potential inhibitory effect when docked against the sequence of the amino acids of the protein targets of (PDB: 6YI3) of the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein which is essential for linking the viral genome to the viral membrane. (Figure S4d), (Figure S4e) In this project for the first time I generated a Comparative Docking Cluster Analysis between the Remdesivir and My prototype chemical library of the Roccustyrna_gs, the Roccustyrna_fr, and the Gissitorviffirna_TM small molecules in combination with the [amino ({4 ? [(2R,3R) ? 2 ? [(2S) ? 3 ? { [(1S,2S) ? 1 ? { [(S) ? 1,3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1,7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium peptide mimetic scaffolding when docked onto the SARS-COV-2 protein targets of (PDB: 7bv2) with the averaged docking energy values of (Num_Members: 40, Total_Energy: 2.103, vdW:-5.122, coulomb:-4.977, internal: 12.203, rmsd: 3.183 and $Number of Clusters: 10, $Seed:-1985, $Leader_Info 1 { Num_Members: 63 Total_Energy:-0.883, vdW:-6.041, coulomb:-7.045, internal: 12.203) KcalMolA respectively. More specifically, in this project I unified generalized Quantum Negative Energies for k-nearest values and Quantum Uncertainities for re-generalizing bosonic formalisms of these k-nearest neighbors of Molecular Pairs (MMP) and von Neumann formulations for Dirac formulation states as a Tipping–Ogilvie and Machine Learning application within this Quantum computing context with algebraic multi-metrics characteristics targeting the atomistic level of the protein apparatus of the SARS-COV-2 viral characteristics.
An Adaptive Weighted KNN Positioning approach through nonlinear electrodynamics to simulate an advanced Quantum mechanical inverse docking algorithm was applied in this unified protocol by providing further insight on a ?neuron (ι), φ D [r2] S [r1] Chern-Simons Topology improver for generating a negative docking energy effect of the highest docking energy values. By performing Euclidean Geometrics driven stationary phase approximations around these [amino ({4 ? [(2R,3R) ? 2 ? [(2S) ? 3 ? { [(1S,2S) ? 1 ? { [(S) ? 1,3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1,7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl} amino) methylidene] azanium pharmacophore merging critical points, I obtained the asymptotic expansions of the Roccustyrna_fr’s merged holomorphic chemical block systems against the specific combination of (PDB: 6mq2), (PDB: 6woj), (PDB: 7khp), (PDB: 7b3d), (PDB: 7b3o), (PDB: 6w23), and (PDB: 6XS6) protein targets (SI Appendix), (SI Appendix II), (SI Appendix III), (SI Appendix IV), (SI Appendix V), (SI Appendix VI), (SI Appendix VII), (SI Appendix VIII), (SI Appendix XII), (SI Appendix XIII), (SI Appendix IX), (SI Appendix XIV), (SI Appendix XV), (SI Appendix XVI), (SI Appendix XVII), (SI Appendix XVIII), (SI Appendix XIX), (SI Appendix XX), (SI Appendix XXI), (SI Appendix XXII), (SI Appendix XXIII), (SI Appendix XXIV), (SI Appendix XXV) (SI Appendix XXVI), (SI Appendix XXVII). (SI Appendix XXVIII), (SI Appendix XXIX), (SI Appendix XXX), (SI Appendix XXXI), (SI Appendix XXXII), (SI Appendix XXXIII), (SI Appendix XXXIV), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XVIII), and ((Cluster Docking Energy Table1), (SI Appendix I), (Cluster Docking Energy Table2), SI Appendix I), (Docking Energy Table1), (SI Appendix I), (Docking Energy Table2), (SI Appendix I), (Docking Energy Table3), (SI Appendix I)). (Illustration1)
This annotated cluster of Druggable Scaffolds were able of producing the highest rates of Negative Docking Energy scoring values when virtually combined with the Amprenavir, Asunaprevir, Atazanavir, Boceprevir, Cytarabine, Darunavir, ritonavir, Sorivudine, Taribavirin, Tenofovir, Valganciclovir, Vidarabine, Lopinavir, Sofosbuvir, Zanamivir, Penciclovir, Nelfinavir, Merimepodib, Maribavir, Indinavir, Inarigivir, Galidesivir, Famciclovir, and Faldaprevir FDA approved antiviral drugs against the SARS-COV-2 protein binding sites of the (PDB: 6M2Q) SARS-CoV-2 3CL protease (3CL pro) apo structure (space group C21) inside the amino acid sequence oV-M-ARG-4, v-S-ARG-4, v-S-MET-6, v-M-ALA-7, v-S-PHE-8, v-M-GLY-11, v-M-LYS-12, v-S-LYS-12, v-M-GLU-14, v-S-GLU-14, v-M-GLY-15, v-M-THR-24, v-S-THR-24, v-M-THR-25, v-S-THR-25, v-M-THR-26, v-S-THR-26, v-M-VAL-35, v-S-VAL-35, v-S-ARG-40, v-S-HIS-41, v-M-THR-45, v-M-SEr-46, v-S-SEr-46, v-S-MET-49, v-M-ASN-53, v-S-ASN-53, v-S-TYR-54, v-M-ALA-70, v-M-GLY-71, v-M-ASN-95, v-S-LYS-97, v-M-PRO-99, v-S-LYS-102, v-S-VAL-104, v-M-ILE-106, v-S-GLN-107, v-M-PRO-108, v-M-GLY-109, v-S-GLN-110, v-M-THR-111, v-S-ASN-119, v-M-GLY-124, v-S-TYR-126, v-M-GLN-127, v-M-CYS-128, v-S-ARG-131, v-S-LYS-137, v-M-LEU-141, v-M-ASN-142, v-S-ASN-142, v-M-GLY-143, v-M-ASN-151, v-S-ASN-151, v-M-ILE-152, v-M-ASP-153, v-S-ASP-153, v-S-SEr-158, v-M-MET-165, v-S-MET-165, v-M-GLU-166, v-S-GLU-166, v-M-LEU-167, v-S-PRO-168, v-M-GLU-178, v-M-VAL-186, v-S-VAL-186, v-S-ARG-188, v-M-GLN-189, v-S-GLN-189, v-M-THR-190, v-S-TRP-218, v-M-LEU-220, v-M-ASN-221, v-S-PHE-223, v-M-TYR-237, v-S-TYR-237, v-S-TYR-239, v-M-ASP-245, v-S-ASP-245, v-S-HIS-246, v-S-ILE-249, v-M-GLU-270, v-S-GLU-270, v-S-LEU-271, v-M-LEU-272, v-M-GLN-273, v-M-ASN-274, v-S-ASN-274, v-M-GLY-275, v-M-MET-276, v-M-ASN-277, v-S-ASN-277, v-M-GLY-278, v-M-LEU-286, v-S-LEU-286, v-M-LEU-287, v-S-LEU-287, v-S-ASP-289, v-S-GLU-290, v-S-THR-292, v-S-PRO-293, v-M-PHE-294, v-S-PHE-294, v-S-ARG-298, v-M-GLN-299, v-S-GLN-299, v-M-GLY-302, v-M-VAL-303, v-M-PHE-305 (SI Appendix I), (SI Appendix II), (SI Appendix III), (SI Appendix IV), (SI Appendix V), (SI Appendix VI), (SI Appendix VII), (SI Appendix VIII), (SI Appendix XII), (SI Appendix XIII), (SI Appendix IX), (SI Appendix XIV), (SI Appendix XV), (SI Appendix XVI), (SI Appendix XVII), (SI Appendix XVIII), (SI Appendix XIX), (SI Appendix XX), (SI Appendix XXI), (SI Appendix XXII), (SI Appendix XXIII), (SI Appendix XXIV), (SI Appendix XXV) (SI Appendix XXVI), (SI Appendix XXVII). (SI Appendix XXVIII), (SI Appendix XXIX), (SI Appendix XXX), (SI Appendix XXXI), (SI Appendix XXXII), (SI Appendix XXXIII), (SI Appendix XXXIV), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), (SI Appendix XXXV), (SI Appendix XXXVI), (SI Appendix XXXVII), and (SI Appendix XVIII), ((Cluster Docking Energy Table1), (SI Appendix I), (Cluster Docking Energy Table2), SI Appendix I), (Docking Energy Table1), (SI Appendix I), (Docking Energy Table2), (SI Appendix I), (Docking Energy Table3), (SI Appendix I)). Additionally, the same combination of My drug design novelties interacted with the highest docking energy values onto the binding sites of the (PDB: 6WOJ) protein targets of the SARS-CoV-2 macrodomain (NSP3) in complex with ADP-ribose of the targeting sequence of V-M-ALA-21, v-M-ASP-22, v-S-ASP-22, v-M-GLU-25, v-S-GLU-25, v-M-ALA-38, v-M-ALA-39, v-S-ASN-40, v-M-TYR-42, v-M-GLY-46, v-M-GLY-47, v-M-GLY-48, v-M-VAL-49, v-S-VAL-49, v-M-ALA-50, v-M-GLY-51, v-M-ALA-52, v-S-LEU-53, v-M-VAL-95, v-S-VAL-95, v-M-VAL-96, v-M-PRO-98, v-S-ASN-101, v-S-LEU-109, v-S-PRO-125, v-M-LEU-126, v-S-LEU-126, v-M-SEr-128, v-M-ALA-129, v-M-GLY-130, v-M-ILE-131, v-S-ILE-131, v-S-PHE-132, v-M-GLY-133, v-M-ALA-134, v-S-PRO-136, v-M-SEr-139, v-M-ALA-154, v-M-VAL-155, v-S-VAL-155, v-M-PHE-156, v-S-PHE-156, v-M-ASP-157, v-M-LEU-160, v-S-LEU-160, v-M-GLU-120 amino acids respectively when compared with Amprenavir, Asunaprevir, Atazanavir, Boceprevir, Cytarabine, Darunavir, ritonavir, Sorivudine, Taribavirin, Tenofovir, Valganciclovir, Vidarabine, Lopinavir, Sofosbuvir, Zanamivir, Penciclovir, Nelfinavir, Merimepodib, Maribavir, Indinavir, Inarigivir, Galidesivir, Famciclovir, and Faldaprevir FDA approved antiviral drugs while targeting the (PDB: 7khp) (Figure S9A), (PDB: 6WOJ) (Figure S9B), (PDB: 7B3D) (Figure S9C), (PDB: 6M2Q) Figure S9D), (PDB: 6lu7) (Figure S9E), (PDB: 6wzu) (Figure S9F), (PDB: 1XU9) (Figure S9G), (PDB: 3TWU) (Figure S9H), (PDB: 7BEO) (Figure S9I), (PDB: 1XAK) (Figure S9J) protein targets. More precisely, I generally made use of the vector field ∂/∂t, which is globally well defined, as the kernel of dz and dz at each point can be viewed as a topological theory of class H.
A Cluster Comparative Docking Analysis between Gisitorviffirna_TM, Roccustyrna_gs, Roccustyrna_fr, DRVYIHPFXmimetic drug designs and the FDA Molnupiravir and Nirmatrelvir, and the Antihypertensive Drugs of Candesartan, Telmisartan, Losartan, Olmesartan, and the Valsartan Drug inside the angiotensin receptor type 1 (AT1R) protein targets.
Ιn this effort Quantum Energy Negativities extracted from the above Quantum Communication Systems ((Supplement Material) Equation Master1-13), ((Iconics1-6), ((Eqs1-400), (Iconics1-4) Supplementary Material METHODS AND MATERIALS)), and (Eqs1-325)) that use entanglement in various ways and for different purposes in a reducible manner generated the apparent loss of degrees of freedom of the original theory in terms of Quantum information in a reduced one through the use of QFT’s internal Quantum network. These translations have been achieved by generalizing Hidden Gene to Protein Interactions of the Hidden Pharmacophoric Subgroups from two oral antivirals, the Molnupiravir drug and Nirmatrelvir-Ritonavir drugs, and the Antihypertensive Drugs of Candesartan, Telmisartan, Losartan, Olmesartan, and the Valsartan Drug which have been used as an outpatient treatment of mild to moderate COVID-19 patients who are at risk for progression. A new Quantum crypto-metalanguage approach of the Quantum simulation of QFT negative reductions of degrees of infinite freedom is proposed here to quantize CS operators from AT1Rs blockers required for ACE2 endocytosis in SARS-CoV-2 infection which is adjusted before being simulated and are strongly relating to how one clustered pharmacophoric element occupies the same Quantum energy phase in an alternative XYZ coordinated smile system for the fragmentizing and the re-merging of the chemical structures of the angiotensin receptor type 1 (AT1R) semi-negative inhibitors of the Candesartan, Telmisartan, Losartan, Olmesartan, and the Valsartan into new Drug Designs with increased levels of Negative Docking Energy Values. More specifically the Antihypertensive Drugs of the Candesartan Conformer of 3D_ CID _ 2541_4ea3_ 637677dbbeFFFTˆUµ (x) superimposed states targets the (PDB: 4ea3) binding domains with semi-negative docking values (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) of (1,3e5dd31abd lig and 1, -5.747, 84.926, 6.790, -29.284) KcalMolA with positive Total Energies of (84.926) KcalMolA due to lack of the so called completely negative superoperator and its natural extension to the tensor product. In addition the Telmisartan Conformer3D_ CID _65999_4ea3_ 6376777d59f4e hits into the same binding cavities with positive Total Energies of (110.429) KcalMolA with the Docking parameters of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) and Docking scoring values of (1,6375f1a51d lig and 1, -6.026,110.429, 3.919, -20.915) KcalMolA because of its complete positivity characterizing such a non-Triangularized Quantum instrument-like pharmacophore. The same sufficient condition cannot be physically realizable also for the Losartan Conformer3D_ CID_3961_4ea3_ 637675d2e4360 since it is observed that the chemical structure of this antihypertensive drug binds into the (PDB: 4EA3) protein targets with a semi-inhibitory docking effect while generating some of positive Total Energies of (92.247) KcalMolA and related docking outputs of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1,7b462582c3 lig and 1, -6.058,92.247, 0.346, -20.606) KcalMolA. The Olmesartan Conformer3D_ CID _ 158781_4ea3_ 637675667c4a3 also produced positive Total Binding Energies of 87.923KcalMolA with docking properties of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) when docked onto the (PDB:4ea3) binding residues with (1, f0e83c3c08 lig and 1, -5.863, 87.923, 8.122, -33.252) KcalMolA implying that this Drug represent a positive super operator, so that Olmesartan’s Total Binding Energy is completely positive against all the above mentioned PDB IDs and relative protein targets. Similarly, any non-Triangularized Total Energy measuring system should have its extended measuring system for any realizable docking system. The (PDB: 6lzg) binding cavities were also covered by the FDA approved Drugs of the Valsartan, Candesartan, Losartan, and Olmesartan but with positive Total Energies unfortunately indicating the fact that these Drugs weren’t able of blockading the core element of the (PDB: 6lzg) protein targets at least computationally. In detail the Candesartan Conformer3D_CID_ 2541_6lzg_ 637673b72423a targeted the (PDB: 6lzg) binding domains with a positive docking energy effect of positive Total Energies of 84.310KcalMolA with binding affinity values (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) of (1, b4c7c9b174 ligand 1, -7.309, 84.310, -12.684, -10.784) KcalMolA. The Losartan Conformer3D_ CID_3961_6lzg_ 637672e4b963e hitted the (PDB: 6lzg) Protein Targets also with positive Total Energies of 84.310KcalMolA and other parameters of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1,6ab04e92d lig and 1, -7.125, 87.761, -15.975, -7.183) KcalMolA. The Olmesartan Conformer3D_ CID _158781_6lzg_ 6376722b32225 binds onto the (PDB: 6lzg) binding residues with a semi positive inhibitory effect of positive Total Energies of 88.260KcalMolA and binding affinity values of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1,8edd42d737 ligand 1, -6.570, 88.260, -6.692, -18.765) KcalMolA. The Candesartan’s Conformer3D_ CID _2541_4ea3_ 637677dbbeFFFTˆUµ (x) produced a semi negative docking effect with positive Total Energy Values of 84.926 KcalMolA and binding affinity energies of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1,3e5dd31abd lig and 1, -5.747, 84.926, 6.790, -29.284) KcalMolA. Finally, the Telmisartan’s Conformer3D_ CID_65999_ 4ea3_ 6376777d59f4e interacted with an intermediate negative docking affinities with positive Total Energies of 110.429KcalMoLA and relative DockThor binding affinity scoring values of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1,6375f1a51d lig and 1, -6.026,110.429,3.919, -20.915) KcalMolA. At the same time the Losartan’s Conformer3D_ CID_3961_4ea3_ 637675d2e4360 produced some of (92.247) KcalMolA non-negative binding Total Energies. The same type of an in-silico semi inhibitory effect also observed by the Olmesartan’s Conformer3D_ CID_158781_ 4ea3_637675667c4a3 when approached the (PDB: 4ea3) protein targets with positive Total Energies of 87.923KcalMolA and generated comparative docking outputs (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) of (1, f0e83c3c08 lig and 1, -5.863,87.923,8.122, -33.252) KcalMolA. The PAXLOVID (Nirmatrelvir) 63737f0dbf777 ligand conformer generated a partial inhibitory effect with positive Total Energy Docking Values of (26.299) KcalMoLA and other less negative vdW and Elec. Energy scoring values of (Table Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1, -7.304,26.299, -10.888, -11.924 run 8, -7.304,26.299, -10.888, -11.924, run 5, -7.570,28.015, -14.907, -6.462, run 9, -7.791,28.022, -17.748, -3.806) KcalMolA. The Molnupiravir b4dac186 ligand conformer generated some of low negative binding affinity scoring values with positive Total Free Energies of (6.750) KcalMolA and other binding scoring values of (1, -6.953,6.394, -11.120, -16.458, run 3, -6.953,6.394, -11.120, -16.458, run 7, -6.919,6.750, -9.433, -18.712, run 2, -6.891,8.520, -11.763, -15.567) KcalMolA. On the other hand, the DRVYIHPFXmimetic X_4ea3X_63887d6da75a7 Drug design interacted inside the same (PDB: 4ea3) Protein Targets only with negative Total Energies, and vdW and Elec. Energies (Rank, File, ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) of (1, abdd7947 Lig and 1, -6.221, -25.657,2.768, -24.410) KcalMolA. (2,5-42) I have illustrated at this point the power of such a Flexible Heuristic Algorithmic Approach as illustrated in (BIOGENEA.Quantum Function1) and interpreted in a distinct Quantum circuit where LnAdS5 refers to qubit preparations, and certain 1-and 2-N qudit gates for automatic molecule fragmentation as a meaningful application for translating these complex physical-like devices interacting with virologic functions. That shows the result of AdS5 Quantum fields Theory (QFT) Reduction measurement where M↓ represent the states of the Generalized Chemical Block Systems and the operator representing the interaction-dynamics, the docking system that measures the Quantum Negative Docking Energy outputs as refined by the BIOGENEA (QCS) - (QEN) s apparatus. After this an observer can finally see Hidden Entanglement Negativity Translations and Uncertainty Quantum Relationships revealed after comparing the docking outputs of the Roccustyrna and Gissitorviffirna Drug Designs with Molnupiravir and Nirmatrelvir Oral COVID19 Antiviral Drugs.
To obtain docking insights into RORγ, I have determined the first crystal structure of a ternary complex containing RORγ ligand-binding domain (LBD) bound with the RoccustyrnaTM_gs_convs_ 2.617493ae06 inhibitors in combination with the repressor peptide, a 22-mer peptide from silencing mediator of retinoic acid and thyroid hormone receptor (SMRT). This 22-mer peptide mimetic ligand [amino ({4 ? [(2R,3R) ? 2 ? [(2S) ? 3 ? { [(1S,2S) ? 1 ? { [(S) ? 1,3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1,7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium generated also negative docking energy values with a potential inhibitory effect when docked against the (PDB: 4ea3) structure of the N/OFQ Opioid Receptor in Complex with a Peptide Mimetic Members of the opioid receptor family of the G-protein-coupled receptors (GPCRs) which have been found throughout the peripheral and central nervous system and have key roles in analgesia.
These NP-peptide mimetics computationally interacted with the GissitorviffirnaTM9 (6R) ? 6 ? [(3S) ? 2 ? [(1Z) ? amino ({1H ? 1,3 ? benzodiazol ? 2 ? yl [(3R) ? 3 ? ethyloxolan ? 3 ? yl] methylidene}) ? λ? ? phosphanyl] ? 5 ? sulfanylidene ? 1,2,4 ? triazolidin ? 3 ? yl] ? 4 ? oxa ? 1 ? azabicyclo [3.1.0] hexane ? 3 ? thione ligand inside the Interacting chains A and constructed Hydrophobic Interactions (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST, LIGCARBONIDX, PROTCARBONIDX, LIGCOO, PROTCOO) of (127, iLE, A, 1501, 0NN, A, 3.80, 5034, 611, 6.900, 59.959, 3.464, 3.456, 58.734, 4.496, 131, TYR, A, 1501, 0NN, A, 3.47, 5011, 641, 7.105, 52.254, 3.207, 4.723, 54.677, 2.522, 131, TYR, A, 1501, 0NN, A, 3.92, 5032, 644, 7.291, 57.619, 2.710, 5.233, 56.289, 0.349, 134, mET, A, 1501, 0NN, A, 3.21, 5011, 671, 7.105, 52.254, 3.207, 7.459, 49.203, 2.269, 135, PHE, A, 1501, 0NN, A, 3.66, 5011, 684, 7.105, 52.254, 3.207, 4.196, 51.520, 5.303, 219, iLE, A, 1501, 0NN, A, 3.40, 5010, 1247, 7.964, 52.247, 4.430, 7.257, 54.433, 6.935, 276, tRP, A, 1501, 0NN, A, 3.83, 5018, 1691, 10.228, 52.693, 0.305, 12.965, 50.175, 1.202) KcalMolA, hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (107, GLN, A, 1501, 0NN, A, True, 2.32, 3.15, 142.09, false, 5027, nam, 460, o2, 10.643, 57.235, 6.108, 10.149, 54.857, 8.120) KcalMolA, salt Bridges (RESNR, RESTYPE, RESCHAIN, PROT_IDX_LIST, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST, PROTISPOS, LIG_GROUP, LIG_IDX_LIST, LIGCOO, PROTCOO) of (130, ASP, A, 633, 634, 1501, 0NN, A, 2.95, false, Tertamine, 5016, 10.495, 54.509, 2.009, 7.967, 53.234, 2.822) KcalMolA, within the 0NN:B:1501 (0NN) binding domains.
By performing stationary phase approximations on and between the chemical spaces of these NP-peptide mimetic a Hermitian operator is generalized to describe the Quantum Negative Meter Observables of the BIOGENEA (QEN) s apparatus. In this measurement model, the Hilbert space describes the Negative Energy States, the unitary operator H describes the time-evolution of the composite system Μ, S, and the density operator σ describes the generalized chemical spaces around the pharmacophore merging critical points as obtained by the asymptotic expansions of the [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium against the nociceptin/orphanin FQ (N/OFQ) peptide receptor (NOP, also known as ORL-1) which was discovered relatively recently by molecular cloning and characterization of an orphan GPCR and has a markedly distinct pharmacology, featuring activation by the endogenous peptide N/OFQ, and a unique selectivity for exogenous ligands. This NP-peptide mimetic ligand that represents the projector onto the subspace of that system’s state is mathematically represented by a density operator.
Then the probability to get the answer is given by the Born rule corresponding to the eigenvalues of the interacted chemical spaces inside its Interacting chains B while generating Hydrophobic Interactions (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST, LIGCARBONIDX, PROTCARBONIDX, LIGCOO, PROTCOO) of (110, ASP, B, 1501, 0NN, B, 3.64, 5093, 3404, 15.850, 36.473, 30.032, 15.376, 35.646, 26.515, 127, iLE, B, 1501, 0NN, B, 3.61, 5101, 3538, 12.580, 36.226, 35.455, 9.104, 37.184, 35.216, 131, TYR, B, 1501, 0NN, B, 3.51, 5078, 3568, 13.188, 43.376, 42.938, 10.614, 41.084, 42.268, 134, mET, B, 1501, 0NN, B, 3.30, 5078, 3598, 13.188, 43.376, 42.938, 13.442, 46.541, 42.054, 135, PHE, B, 1501, 0NN, B, 3.78, 5078, 3611, 13.188, 43.376, 42.938, 10.230, 44.214, 45.129, 219, iLE, B, 1501, 0NN, B, 3.19, 5077, 4193, 14.006, 43.126, 44.162, 13.306, 41.235, 46.635) KcalMolA, hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (107, GLN, B, 1501, 0NN, B, True, 2.17, 3.03, 145.56, false, 5094, nam, 3387, o2, 16.420, 38.706, 33.233, 15.800, 41.083, 31.453) KcalMolA, and Salt Bridges (RESNR, RESTYPE, RESCHAIN, PROT_IDX_LIST, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST, PROTISPOS, LIG_GROUP, LIG_IDX_LIST, LIGCOO, PROTCOO) of (110, ASP, B, 3406, 3407, 1501, 0NN, B, 5.15, false, Tertamine, 5103, 14.303, 37.179, 31.615, 15.465, 33.941, 27.776, 130, ASP, B, 3560, 3561, 1501, 0NN, B, 3.03, false, Tertamine, 5083, 16.264, 41.067, 37.553, 13.753, 42.618, 36.849) KcalMolA within the OLA:A:1503 (OLA) binding domains when combined at least virtually with the GissitorviffirnaTM8 {1 ? [(R) ? [(1S, 3R) ? 3 ? [(R) ? amino (carbamothioylamino) methyl] diaziridin ? 1 ? yl] ({6 ? oxo ? 2 ? [(2S, 5R) ? 3, 4, 5 ? trifluoro ? 2, 5 ? dihydrofuran ? 2 ? yl] ? 6, 7 ? dihydro ? 1H ? purin ? 8 ? yl}amino) phosphoroso] ? 1H ? azirin ? 2 ? yl}thiourea druggable pharmacophoric compounds according to the projection postulate since the post-measurement state is obtained via each molecular complex state-transformation. Here, the observable-operator and its spectral decomposition uniquely determine the feedback Quantum State Negativity Transformations for Negative Docking Energy Outcomes by revealing atomic details of these ligand-receptor recognitions and selectivities. This NP-Peptide mimetic Compound mimics the first four amino-terminal residues of the NOP-selective peptide antagonist UFP-101, a close derivative of N/OFQ, and provides important clues to the binding of these peptides against the Interacting chains A, B when generating Hydrophobic Interactions (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST, LIGCARBONIDX, PROTCARBONIDX, LIGCOO, PROTCOO) of (55, VAL, A, 1503, oLA, A, 3.14, 5076, 61, 12.465, 54.202, 19.220, 15.257, 53.858, 17.827, 74, TYR, B, 1503, oLA, A, 3.70, 5063, 3120, 6.092, 66.908, 21.039, 4.974, 67.226, 24.547, 74, TYR, B, 1503, oLA, A, 3.84, 5065, 3118, 7.494, 65.194, 22.260, 5.480, 66.235, 25.364, 109, THR, A, 1503, oLA, A, 3.63, 5073, 472, 10.848, 56.661, 21.596, 9.256, 56.761, 18.335, 112, LEU, A, 1503, oLA, A, 3.47, 5074, 495, 12.017, 56.000, 20.882, 14.754, 57.769, 19.695, 113, LEU, A, 1503, oLA, A, 3.87, 5066, 503, 7.687, 63.702, 22.516, 9.534, 62.781, 19.239, 113, LEU, A, 1503, oLA, A, 3.61, 5069, 504, 9.319, 61.337, 22.560, 11.679, 63.141, 20.513, 115, PHE, A, 1503, oLA, A, 3.69, 5061, 517, 4.783, 68.177, 19.295, 7.499, 66.406, 17.540, 115, PHE, A, 1503, oLA, A, 3.50, 5063, 519, 6.092, 66.908, 21.039, 8.474, 66.519, 18.510, 117, PRO, A, 1503, oLA, A, 3.68, 5064, 538, 6.405, 65.426, 21.218, 5.582, 63.725, 18.059) KcalMolA against the OLB:A:1502, (OLB) binding cavities. These structures also show substantial conformational differences in the pocket regions between NOP and the classical opioid receptors κ (ref.5) and μ (ref.6), and these are probably due to a small number of residues that vary between these receptors. This NP-mimetic structure explains the divergent selectivity profile of NP and provides a new structural template for the design of NP mimetic ligands that are capable of generating Hydrophobic Interactions (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST, LIGCARBONIDX, PROTCARBONIDX, LIGCOO, PROTCOO) of (139, PHE, A, 1502, oLB, A, 3.86, 5056, 712, 1.882, 42.380, 11.862, 3.617, 44.197, 8.925, 143, ALA, A, 1502, oLB, A, 3.87, 5043, 743, 0.721, 36.187, 11.552, 2.432, 36.184, 8.080, 177, LEU, A, 1502, oLB, A, 3.88, 5055, 946, 0.935, 41.513, 11.040, 1.583, 44.430, 10.614) KcalMolA, hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (89, TYR, A, 1502, oLB, A, True, 1.50, 2.37, 151.58, true, 320, o3, 5053, o3, 0.659, 30.781, 8.969, 0.323, 32.597, 7.489) KcalMolA inside the OLC:B:1502, (OLC) targeted binding residues of the Interacting chains B, while constructing Hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (147, ASP, B, 1502, oLC, B, True, 1.69, 2.58, 155.79, true, 3698, o3, 5123, o3, 6.436, 65.958, 49.872, 8.468, 67.082, 48.754) KcalMolA. On the other hand this innovative Drug Design Conformer, the GissitorviffirnaTM-d5dc2541d4 ligand generated an in-silico blocking effect only interacted with Negative Total Free Energy Docking values of (1, -6.725, -38.697, -6.406, -35.072, run 12, -6.725, -38.697, -6.406, -35.072, run 12, -6.918, -38.398, -16.867, -17.322, run 8, -6.637, -37.566, -9.611, -22.509) KcalMolA inside the (PDB: 6wzu) protein targets. At the same time a semi-negative docking effect was detected from the Nirmatrelvir compound with positive Total Energy Interaction docking values (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) of (1, PAXLOVID (Nirmatrelvir) e92b8739e1 ligand of (1, -5.957, 49.082, -0.000, -0.001, run 1, -5.957, 49.082, -0.000, -0.001, run 1, -5.957, 49.082, -0.000, -0.001, run 1, -5.957, 49.082, -0.000, -0.001) KcalMolA when combined with the Molnupiravira0932ea8a7 ligand against the same binding domains. More precisely the Molnupiravir conformer interacted with Positive Total Energy Interactions of (1, -5.428, 32.769, -0.000, -0.003, run 2, -5.428, 32.769, -0.000, -0.003, run 2, -5.428, 32.769, -0.000, -0.003, run 2, -5.428, 32.769, -0.000, -0.003) KcalMolA due to the missing of the (1S, 3S, 7R) ? 7 ? amino ? 1 ? [(R) ? {3 ? sulfanylidene ? 2, 4, 6 ? triazabicyclo [3.1.0] hexa ? 1, 4 ? dien ? 6 ? yl} [(E) ? 2 ? [(3R) ? 3 ? [(2R, 5R) ? 3, 4, 5 ? trifluoro ? 2, 5 ? dihydrofuran ? 2 ? yl] ? 3H ? 1, 2, 4 ? triazol ? 5 ? yl] diazen ? 1 ? yl] phosphoroso] ? 1, 2, 4, 6 ? tetraazaspiro [2.4] heptane ? 5 ? thione pharmacophoric substitutions. In addition, the GissitorviffirnaTM8ac33ef19 ligand conformers of (1, -4.598, -7.504, -0.000, 0.076, run 10, -4.598, -7.504, -0.000, 0.076, run 10, -4.598, -7.504, -0.000, 0.076, run 10, -4.598, -7.504, -0.000, 0.076) KcalMolA. The Molnupiravir_5vyh_ 6372632b98921 ligand conformer generated an intermediate inhibitory outcome inside the (PDB: 5vyh) binding domains since it was interacted with positive Total Energies of (34.068) KcalMolA and docking activities of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1, molnupiravirac1e1b4022 lig and 1, -1.667, 34.068, -0.000, -0.013) KcalMolA due to the lack of the 4 ? [3 ? (aminomethyl) oxiren ? 2 ? yl] ? 4, 5 ? diaza ? 2λ? ? phosphaspiro [bicyclo [3.1.0] hexane ? 2, 2' ? oxaphosphirane] ? 3 ? thione druggable scaffoldings. The same partial inhibitory effect was observed when the Molnupiravir_6lu7_ 637265a213f4d ligand conformer generated a partial negative docking effect of less positive docking energy values when docked into the (PDB: 6lu7) protein targets with the binding affinity scoring values of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1, molnupiravir7b59c1 lig and 1, -5.428, 34.075, -0.000, -0.005) KcalMolA due to the absence of the (1S, 5S) ? 4 ? [(2R, 3S) ? 3 ? [(3R) ? 2 ? amino ? 1 ? fluoro ? 5 ? sulfanylidene ? 3H ? 1, 2, 4 ? triazol ? 3 ? yl] oxiran ? 2 ? yl] ? 4, 5, 6 ? triaza ? 2λ? ? phosphaspiro [bicyclo [3.1.0] hexane ? 2, 2' ? oxaphosphirane] ? 3 ? thione substituted ligand. The Molnupiravir_1xak_ 637265fc11b93 ligand also interacted with intermediate docking affinities and positive Total Energies against the (PDB: 1xak) Protein Targets with the scoring values of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with (1, molnupiravir 531711241a lig and 1, -5.591, 12.564, 4.722, -29.328) KcalMolA because of the missing pharmacophoric groups of the (Z) ? 4 ? amino ? N ? [(1Z) ? amino-methylidene] ? N' ? [(Z) ? 2 ? {6 ? [(1Z) ? [(fluoromethyl) imino] methyl] ? 3 ? sulfanylidene ? 1, 2, 4 ? triazabicyclo [3.1.0] hex ? 2 ? en ? 6 ? yl} ? N' ? methyl-ethanimidamido] ? 2 ? oxobutanimidamide chemical element. At the same time the Molnupiravir_5rgw_ 63726ae391d65 ligand generated again positive Total Energies when interacted with the SARS-CoV-2 protein targets of (PDB:1xak) with the docking values of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with some of (1, molnupiravird26f2bece9 lig and 1, -6.867, 7.794, -7.938, -19.751) KcalMolA due to missing pharmacophoric elements of [2 ? (aminomethyl) ? 2 ? { [(2E) ? 3 ? oxofuran ? 2 ? ylidene] methyl} ? 2λ? ? azaphosphiridin ? 1 ? yl] amino (1R, 4S) ? 3, 3 ? dimethyl ? 6 ? oxo ? 2λ? ? thia ? 5 ? azaspiro [bicyclo [3.2.0] heptane ? 2, 1' ? thiirane] ? 4 ? carboxylate. The FDA drug of the Molnupiravir_5rf6_ 63726b4acf0fd ligand conformer generated finally positive Total Energies of 7.385KcalMolA with the docking values of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with some of (1, molnupiravir28276be760 lig and 1, -7.051, 7.385, -12.069, -16.022) KcalMolA while co-interacting with the Valsartan_ 1y8jX_6348460bb635e ligand conformer for the generation again of an intermediate docking effect with positive docking values of 73.198KcalMolA and observed docking parameters of (Table, Rank, File ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) with some of (1, valsartan ea8754f6fa lig and 1, -7.005, 73.198, -10.604, -20.20) KcalMolA. That Positive Docking Energy effect produced from the combination of the Molnupiravir and Valsartan Drugs means that these two chemical structures weren’t able of generalizing the Quantum Negative state Transformations generated by docking measurements’ feedback due to the missing of the Triangularized pharmacophoric group of N ? { [(2S) ? 3 ? (aminomethyl) ? 2 ? [(1R, 2S) ? 2 ? methyldiaziridin ? 1 ? yl] ? 1, 3 ? diazetidin ? 1 ? yl] ({ [(E) ? { [(E) ? (aminomethylidene) amino] methylidene} amino] amino}) aziridin ? 1 ? yl ? λ? ? phosphanyl} ? 3 ? {5 ? [(1E) ? [2 ? (aminomethyl) ? 2 ? methylhydrazin ? 1 ? ylidene] methyl] ? 3, 4 ? dihydroxyfuran ? 2 ? yl} ? 2 ? methyl ? 5 ? sulfanylidene ? 1, 2, 3, 4 ? tetrazole ? 1 ? carboxa-diamino ({ [1 ? ({ [(2S, 3R, 4R, 5R) ? 5 ? (3 ? { [(R) ? [(2 ? amino ? 6 ? oxo ? 8, 9 ? dihydro ? 1λ?, 3λ? ? purin ? 9 ? yl) amino] [(6 ? fluoro ? 3H ? 1λ? ? pyrazin ? 2 ? yl) formamido] phosphoryl] carbamoyl} ? 1, 5 ? dihydro ? 1, 2, 4 ? triazol ? 1 ? yl) ? 3, 4 ? dihydroxyoxolan ? 2 ? yl] methoxy} (cyano) amino) ethenyl] imino}) methanium derivatives. On the hand my DRVYIHPFXmimetic_ 6gid_63887f917804a Drug Design interacted solely with negative docking energy values (Rank, File, ID, Compound, Affinity, Total Energy, vdW Energy, Elec. Energy) of (1, 8f357f6ad2 Lig and 1, -7.178, -36.112, -14.305, -17.511) KcalMolA inside the same (PDB: 6gid). This DRVYIHPFX mimetic ligand generated once more noncovalent interactions for (PDB: 6gid) protein structure of the substrate-free human neprilysin Neprilysin inside it’s transmembrane M13 zinc metalloprotease binding sites which are responsible for the degradation of several biologically active peptides including insulin, enkephalin, substance P, bradykinin, endothelin-1, neurotensin and amyloid-β while targeting the EDO:A:806 (EDO) binding sites when conjoined with the GissitorviffirnaTM7 (1S, 3S, 7R) ? 7 ? amino ? 1 ? [(R) ? [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methyl propyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium {3 ? sulfanylidene ? 2, 4, 6 ? triazabicyclo [3.1.0] hexa ? 1, 4 ? dien ? 6 ? yl} [(E) ? 2 ? [(3R) ? 3 ? [(2R, 5R) ? 3, 4, 5 ? trifluoro ? 2, 5 ? dihydrofuran ? 2 ? yl] ? 3H ? 1, 2, 4 ? triazol ? 5 ? yl] diazen ? 1 ? yl] phosphoroso] ? 1, 2, 4, 6 ? tetraazaspiro [2.4] heptane ? 5 ? thione neo-ligand (SI Appendix XL) within the Interacting chains A. Hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (195, GLY, A, 806, eDO, A, False, 2.75, 3.44, 128.85, false, 5605, o3, 1098, o2, 16.140, 26.425, 9.139, 18.810, 24.341, 8.535, 196, LYS, A, 806, eDO, A, True, 2.29, 3.30, 169.26, true, 1107, n3, 5603, o3, 13.414, 27.389, 11.321, 14.171, 24.687, 13.061, 197, LYS, A, 806, eDO, A, False, 2.11, 3.07, 165.00, true, 1108, nam, 5605, o3, 16.140, 26.425, 9.139, 15.191, 23.894, 7.679, 373, ARG, A, 806, eDO, A, True, 2.49, 2.91, 105.37, true, 2541, ng, 5603, o3, 13.414, 27.389, 11.321, 10.731, 26.906, 12.352) KcalMolA were generated inside the EDO:A:807 (EDO) binding sites when the amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydro-isoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium small molecule conducted with the GissitorviffirnaTM6 (2R, 4R, 5S) ? 2 ? amino ? 3 ? {4 ? amino ? 1 ? [(2R, 3R, 4R, 5R) ? 5 ? [(2R) ? 3 ? (aminomethyl) ? 2 ? (2 ? methyldiaziridin ? 1 ? yl) ? 1, 3 ? diazetidin ? 1 ? yl] ? 3, 4 ? dihydroxyoxolan ? 2 ? yl] ? 5 ? sulfanylidene ? 4, 5 ? dihydro ? 1H ? 1, 2, 4 ? triazole ? 3 ? carbonyl} ? 1 ? [(2S) ? 1 ? [(3R) ? 2 ? amino ? 1 ? methyl ? 5 ? sulfanylidene ? 1, 2, 4 ? triazolidin ? 3 ? yl] propan ? 2 ? yl] ? octahydro ? 1H ? purin ? 6 ? one drug design (SI Appendix XL) inside the same Interacting chains A. Additional Hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (206, THR, A, 807, eDO, A, False, 2.18, 3.02, 143.00, true, 1178, nam, 5609, o3, 22.347, 34.983, 0.162, 23.986, 33.907, 2.135) KcalMolA and water Bridges (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST_A, W, DIST_D, W, DON_ANGLE, WATER_ANGLE, PROTISDON, DONOR_IDX, DONORTYPE, ACCEPTOR_IDX, ACCEPTORTYPE, WATER_IDX, LIGCOO, PROTCOO, WATERCOO) of (346, TYR, A, 807, eDO, A, 3.45, 3.70, 153.83, 117.07, false, 5607, o3, 2314, o3, 5885, 19.543, 35.634, 0.353, 17.673, 37.051, 2.268, 17.274, 38.520, 0.831) KcalMolA inside the EDO:A:808 (EDO) binding domains were constructed when the pharmacophoric scaffold of [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium combined with the GissitorviffirnaTM4 (1S, 5S) ? 4 ? [(2R, 3S) ? 3 ? [(3R) ? 2 ? amino ? 1 ? fluoro ? 5 ? sulfanylidene ? 3H ? 1, 2, 4 ? triazol ? 3 ? yl] oxiran ? 2 ? yl] ? 4, 5, 6 ? triaza ? 2λ? ? phosphaspiro [bicyclo [3.1.0] hexane ? 2, 2' ? oxaphosphirane] ? 3 ? thione small molecule. These novel pharmacophoric designs could receive attention in near for its potential role in modulating blood pressure responses with its inhibition producing an antihypertensive response since to date, several inhibitor bound crystal structures of the human neprilysin extracellular domain have been determined, but a structure free of bound inhibitor or substrate has yet to be reported. Moreover Hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (264, ASP, A, 808, eDO, A, False, 1.87, 2.82, 160.29, true, 1647, nam, 5611, o3, 0.117, 9.051, 23.843, 1.328, 8.662, 26.357) KcalMolA including water Bridges (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST_A, W, DIST_D, W, DON_ANGLE, WATER_ANGLE, PROTISDON, DONOR_IDX, DONORTYPE, ACCEPTOR_IDX, ACCEPTORTYPE, WATER_IDX, LIGCOO, PROTCOO, WATERCOO) of (267, GLN, A, 808, eDO, A, 2.97, 4.00, 153.01, 97.25, true, 1680, nam, 5613, o3, 5702, 1.188, 7.266, 20.727, 3.305, 10.037, 17.948, 0.386, 9.369, 19.348) KcalMolA against the EDO:A:809 (EDO) binding residues were produced when the clustered pharmacophoric element of [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium concerted in these parallel docking experiments with the GissitorviffirnaTM3 (Z) ? 4 ? amino ? N ? [(1Z) ? amino-methylidene] ? N' ? [(Z) ? 2 ? {6 ? [(1Z) ? [(fluoromethyl) imino] methyl] ? 3 ? sulfanylidene ? 1, 2, 4 ? triazabicyclo [3.1.0] hex ? 2 ? en ? 6 ? yl} ? N' ? methylethanimidamido] ? 2 ? oxobutanimidamide ligand (SI Appendix XL) inside the Interacting chains A while Hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (292, ARG, A, 809, eDO, A, True, 3.34, 3.78, 109.41, true, 1876, ng, 5615, o3, 15.143, 37.396, 2.310, 11.668, 36.021, 2.905) KcalMolA and water Bridges (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST_A, W, DIST_D, W, DON_ANGLE, WATER_ANGLE, PROTISDON, DONOR_IDX, DONORTYPE, ACCEPTOR_IDX, ACCEPTORTYPE, WATER_IDX, LIGCOO, PROTCOO, WATERCOO) of (292, ARG, A, 809, eDO, A, 3.92, 3.82, 110.03, 125.26, false, 5617, o3, 1869, o2, 5787, 14.114, 40.643, 3.719, 11.441, 42.049, 0.919, 10.866, 42.346, 4.789) KcalMolA redirected into the EDO:A:810 (EDO) binding domains. Τhe active pharmacophoric element of [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium when combined with the GissitorviffirnaTM1 (2S, 3S) ? 3 ? ({ [(1 ? { [(3R) ? 2 ? (dimethylamino) ? 3 ? [(R) ? hydroxy (2R) ? oxiran ? 2 ? ylmethyl] ? 2λ? ? oxaphosphiran ? 2 ? yl] methyl}hydrazin ? 1 ? yl) methyl] amino} methyl) oxirane ? 2 ? carbonitrile (SI Appendix XL) covered the whole binding surfaces in Interacting chains A while constructing Hydrogen Bonds (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, SIDECHAIN, DIST_H, A, DIST_D, A, DON_ANGLE, PROTISDON, DONORIDX, DONORTYPE, ACCEPTORIDX, ACCEPTORTYPE, LIGCOO, PROTCOO) of (448, GLU, A, 810, eDO, A, False, 3.25, 3.71, 110.69, true, 3124, nam, 5621, o3, 37.123, 59.496, 3.334, 33.639, 60.589, 3.993) KcalMolA and water Bridges (RESNR, RESTYPE, RESCHAIN, RESNR_LIG, RESTYPE_LIG, RESCHAIN_LIG, DIST_A, W, DIST_D, W, DON_ANGLE, WATER_ANGLE, PROTISDON, DONOR_IDX, DONORTYPE, ACCEPTOR_IDX, ACCEPTORTYPE, WATER_IDX, LIGCOO, PROTCOO, WATERCOO) of (444, ALA, A, 810, eDO, A, 3.98, 3.66, 101.99, 89.29, true, 3091, nam, 5619, o3, 5675, 38.720, 56.570, 3.135, 37.118, 57.120, 7.660, 39.164, 54.352, 6.410) KcalMolA inside the EDO:A:811 (EDO) binding sites in conjunction with the [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium Interacting chains GOL:A:817, (GOL), [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium Interacting chains GOL:A:817, (GOL), [amino ({4 ? [(2R, 3R) ? 2 ? [(2S) ? 3 ? { [(1S, 2S) ? 1 ? { [(S) ? 1, 3 ? dihydroisoindole ? 2 ? carbonyl [(2 ? methyl ? 6 ? oxo ? 1, 7 ? dihydropurin ? 8 ? yl) methyl] phosphanyl] carbamoyl} ? 2 ? methylbutyl] amino} ? 2 ? methylpropyl] oxaziridin ? 3 ? yl] butyl}amino) methylidene] azanium when docked in parallel with the Roccustyrna_gs8-1 diamino ({ [1 ? ({ [(2S, 3R, 4R, 5R) ? 5 ? (3 ? { [(R) ? [(2 ? amino ? 6 ? oxo ? 8, 9 ? dihydro ? 1λ?, 3λ? ? purin ? 9 ? yl) amino] [(6 ? fluoro ? 3H ? 1λ? ? pyrazin ? 2 ? yl) formamido] phosphoryl] carbamoyl} ? 1, 5 ? dihydro ? 1, 2, 4 ? triazol ? 1 ? yl) ? 3, 4 ? dihydroxyoxolan ? 2 ? yl] methoxy} (cyano) amino) ethenyl] imino}) methanium neo-ligand (SI Appendix XL) as a first crystal structured inhibitor targeted the extracellular catalytic domain of human neprilysin at 1.9?Å resolution. This structure will provide a reference point for comparisons to future inhibitor substrate bound structures. These neprilysin mimicking structures also revealed that these closed protein-ligand conformations can be adopted in
Discussions
Quantum Homeopathy as Enactment of Double Entangled States points to the fact that some other phenomenon might be operative here, which is the reason why we propose to look at it in terms of an entanglement model along the lines of QFT based Quantum Homeopathy remedies. [1-42] This is how a theoretical ligand reconstruction along those lines might proceed to innovative methods for the generation of 3D structures of potential ligand molecules directly from the 3D structure of the macromolecular binding sites within their binding domains in the form of a 3D graph. [1-48] These drug designing systems and aspects of Quantum Homeopathy Interpretations are still unknown, but their optimization is central for interpreting their entropy signature into many chemical-informatics and bio-informatics tasks, such as sequence alignment, de novo pharmacophore assembly, and phylogenetic tree inference. [1-49] This interpretation of evolutionary Quantum Homeopathies is represented here by the inverse of the effective pharmacophoric size in ultra-low dosages, and could be more general because it is reflecting the degree of stochasticity in this evolutionary process, which depends not only on the effective pharmacophore size, but also from other factors, in particular the transformation of the ultra-small dosage distributions into quantum entropy interactions of druggable scaffolds within SARS-COV-2 protein micro-environments. [1-50] It is interesting to consider how disentanglement of homeopathy remedies and placebo could be represented in terms of a double-slit metaphor in these computational and mathematical chemistry tasks as a strong ritual and a drug designing system of its own by following Turing Machine Rules and hypergeometric quantum functions
Highlights Supplementary
Material,Maths19a,19b,19c,19d,19e,19f,19g,19h,20a,20b,20c,20d,20e,20f1,20f2,20f3,20g,20h,20i,20j,21a,21b,21c,21d,21e,21f,21g,21h,21i,21j,22a,22b,22c,22d,,22e,,22f,22g), and (Figures 1- 133), (OUTPUTs1-3)), (Ic1a), (Ic1b), (Ic1c), (Ic2b), (Ic2c), (Ic2a), (Ic3a), (Ic3b) (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), (Ic3j), ((Iconics4-6), (Supplementary Material METHODS AND MATERIALS)), (Cluster of BIOGENEA_ CONSENSUS_Eqs.1-19), (Supplement Material FUNCTIONS.1 ? 11) according to the similar rule that states in the case of illness where the symptoms of an ill patient have to match the signs which have been observed in healthy volunteers ingesting a certain substance, either in crude form or potentized. [1-58] In this Quantum Homeopathy experiment where the observation (i.e. gaining information) of electrons are the heavy integration of sequence alignment algorithms with water memory tasks led me to diverse drug designing applications from the de novo assembly of whole pharmacophoric fragments [3-59] to the discovery of new drug designs targeted quantitatively to disease trait loci linked phenotypes [3-60], and to the identification and analysis of driver mutations in SARS-COV-2 viral diseases [3-61]. The potentization principle that states that by stepwise diluting and succussing of a substance becomes more active, even beyond Avogadro’s number transition associated finally its wave function [60] with Euclid-equivariant diffusion models by enabling them to leave their source and arrive at the screen as fuzzy sphere like drug designs (Figures1-133), ((Iconics1-4), (Eqs1-400), Supplementary Material METHODS AND MATERIALS)). By learning to denoise a normally distributed set of points throughout this whole drug designing experiment we manage to generate molecule designs from scratch within the binding site of macromolecules. This Sequence alignment OF ZernikeR and Hypergeometric quantum functions for Euclid-equivariant diffusion models that corresponds to the transition from infrabiological entities to biological and chemical ones, as formulated by Szathmary [1-79] following Ganti’s chemoton concept can also be considered as quantum circuit inputs in these Quantum Homeopathy and pharmacophoric generation experiment. According to Ganti, and since life is characterized by the union of features the three essential features of membrane compartmentalization, autocatalytic metabolic network, and informational replicator [80-81] can also be considered as inputs in these Holistic Quantum Functions ((Cluster of BIOGENEA_ CONSENSUS_Eqs.1-20), (Supplement Material FUNCTIONS.1 ? 12)). [3-80] As a result these potentizations of molecules per mol of the anti-viral and ACE2 blockers/substances could also be inserted as non-generalized and parameterized inputs in these Quantum Functions ((Cluster of BIOGENEA_ CONSENSUS_Eqs.1-21), (Supplement Material FUNCTIONS.1 ? 13)) via the same Turing Machine translating process from these potentized remedies according to the simile rule, when, a homeopath uses something which is not present any more. The generated drug designs geometrically mimic the molecular signature of the substances diluted out when treating a symptom picture, which in the past was related to the interference pattern disappearing in ultra-low doses and be replaced by two clear spots in a classic formulation for identifying the global optimum alignment of two sequences. This Quantum Homeopathy experiment involves finding of the lowest weight path through an n × m dynamic programming matrix, where n and m are the lengths of the sequences being compared (it is often the case that n, m) [3-82] in a double-semiotic structure which has not been elaborated on elsewhere [1-87]. ‘Semiotic’ means here that the basic relationship is the one of a semiotic triad developed by Charles S. Peirce [1-88], who pointed out that the semiotic triad of object, sign, and the meaning of the sign is universal. It can be applied to homeopathy in that the homeopathic remedy can be construed as a sign for an object, the substance, conveying a certain meaning, the remedy picture as an approximate solution which is typically constructed by a greedy heuristic using a biologically informed scoring function. These Examples incorporating certain aspects of physics and symmetry into a model tends to increase the accuracy, generalizability, and interpretability of the predictions of the scoring functions including sum-of-pairs, weighted sum-of-pairs and minimum entropy for each of the atomic orbits involved and could imply certain biological assumptions [3-83] when translating the same sense of the symptoms of a disease including the signs of that disease. Both are matched by the law of Similars. There are multiple modes of entanglement present here expecting that this Turing Machine ruled Geometric deep learning research for quantum-based structure-based drug design will follow trends in the pharmaceutical industry: A) The generated drug designs mimics the remedy itself and is something like a magical geometry representation of the past, at least in the case where ultra-high dilutions are used (note that in the case of low dilutions there might be a mix of entangled states and signaling processes via molecules) with a weighted sum-of-pairs, where one may assign different scores to DNA base matches, mismatches, substitutions, insertions and deletions (the scoring system which may also be used to control whether the output alignment is global [3-82] or local [3-84]). Alternatively, for AT1R/ACE2 proteins, a scoring matrix has be used where each ligand-protein complex represents the likelihood that the amino acid in the row will be replaced by the amino acid in the column [3-68,70-85] which is brought into a Euclid special space relationship with a particular diseased organism exhibiting special signs, which in other cases were related to and brought about by this substance, as represented in the knowledge system of homeopathy and its Materia Medica. B) Thus, high-entropy environments promote adaptation, and then success breeds success, that adaptation is most effective in large populations of pharmacophoric fragments which were translated from this Quantum Homeopathy drug design prediction which is at least qualitatively compatible with the available geometry data that are quantitatively testable in this double entanglement situation. The Quantum Homeopathy remedies involved here in an entangled state itself and acts like a phenomenological theory between actual remedy and pharmacophore geometrical data (a). This is achieved when a Quantum Turing Machine generates drug design structures after translating these potentization modern evolutionary theories that include an elaborate and a hidden mathematical description of a quantum hidden macroentanglement into druggable drug designs [12-87] according to our knowledge, where there is a coherent theoretical representation of MTE which can be determined empirically by a statistical analysis of a large protein sequence database on the basis of chemical docking properties between the amino acids and our RoccustyrnaTM drug designs (e.g. polar or non-polar, hydrophilic or hydrophobic). This Quantum Homeopathic Ritual enacts another entangled state between symptomatology of the patient and the remedial substances received following the similarity principle entanglement functions of
Both are matched by the Law of Similars. [35-86] There are multiple modes of entanglement present here: A) The new drug design itself is something like a magical presence of the past, at least in the case where ultra-high dilutions are used (note that in the case of low dilutions there might be a mix of entangled states and signaling processes via small molecule/ligand docking interactions) and directly propose a theoretical framework for turing analysis, in which these quantum Turing Machines are treated as phase transitions, in a technical and physical sense where this transition is the point where two distinct grand potentials are characterizing as units at different levels, such as molecules vs cells (organisms). In this case of the origin of life we put another way, where the transition is from an ensemble of entities at a lower level of organization (for example, small molecules) to an ensemble of higher-level entities (for example, small molecules-ligand complexes).
Concluding Remarks
The exploration of binding affinity prediction methods has a long-standing history. Early studies focused on utilizing empirical formulas or designing handcrafted features coupled with traditional machine learning algorithms for binding affinity prediction [2-187]. In this paper, I have illustrated a quantum partial trace measurement (observation) for geometrically translating a quantum homeopath state into Structure-based drug designs (SBDDs), which is becoming increasingly vital in drug discovery by utilizing the three-dimensional geometry of proteins to identify potential drug candidates. These translations in this project represent a part of a second generalized entangled system which depends on knowledge and technicalities, namely, for solving the correct quantum functions in order to link the remedy and symptom pictures of the patient with hypergeometric shapes. [3-87,109-143] This link has to be sufficiently strong as well as sufficiently correct and similar such that one single global description ensues, namely the remedy picture. The remedy picture that contains symptoms collected in the past and by other subjects. [3-29,35-144] were extracted by the well-known paradoxes of quantum theory, such as Schrödinger's Cat [2-53,54]. These conundra appear if the quantum state of a system is taken as a representation of Quantum Homeopathy Knowledge and what we can know about it, not its presumed objective ‘reality in itself ‘particularly in this process of reduction from QFT to QM which is proved to be much more complex than just the reduction to a finite volume. [2-53,58] In fact, these recent advancements in geometric deep learning, which integrates and processes 3D geometric data, coupled with the QFT’s Hidden Quantum Homeopathy Information from double-blinded and placebo-controlled homeopathic signal transduction that are disentangled into a generalized Quantum Black Hole like paradigm have significantly propelled our structure-based drug design progress especially when some Avogadro Number’s unknown characteristics emerged and several topics involved after translating genomic signatures of clinical samples from patients received homeopathy remedies with viral pneumonia in Wuhan, China. [2-53,54-100] In this paper, we systematically improve the recent progress of geometric deep learning for structure-based drug design targeting a novel peptide signature of AT1R-coronavirus (termed 2019-nCoV) binding domains which is identified [10-111] and is considered among the unknown characteristics of this phylogenetic analysis of 2019-nCoV, as sequenced from nine patients' samples, and has shown that the virus belongs to the subgenus Sarbecovirus. [2-113,114] But it is in investing this state with too much ‘reality’ as an object ‘out there’, independent of our observation of the quantum-computational structure which is intrinsically rooted in QFT, and the quantum-gravitational origin of this same structure where the topics involved, besides those of quantum information and quantum gravity, are non-commutative geometrized (the fuzzy sphere), (Figures1-96), ((Iconics1-4), (Eqs1-400), and Schematic1.(I-VI) Supplementary Material METHODS AND MATERIALS)) and quantum simulated. [2-124] Thus, the computational breaking of macroentanglement in this case of 2019-nCoV which was more similar to two bat-derived coronavirus strains, bat-SL-CoVZC45 and bat-SL-CoVZXC21, than to known human-infecting coronaviruses, including the virus that caused the SARS outbreak of 2003 is a necessary concomitant in this drug SBDD Drug Designing process and of great importance as a possible theoretical support for translating Patient–practitioner–remedy (PPR) entanglement applications between patient, practitioner and homeopathy remedies in these Avogadro Number’s processes of elementary atoms at high energies into negative docking energy values and innovative multi-targeted neoligands. [22-103,54-115] This Entanglement model of homeopathy translations is a quantum hypergeometric example of generalized entanglement predicted by weak quantum theory revealing its greatest strength and promise, not yet touched upon. [2-53,60-116] It has always been an intuition of Wolfgang Pauli, one of the founders of modern QM, that physics will only be complete if it has incorporated consciousness into a final theory of matter [33-120]. Until now, quantum algorithms to simulate QFT [13,14-139] have mainly used lattices. [2-73,100-140] Conversely, this transactional interpretation of quantum mechanics for each task (binding site prediction, binding pose generation, de novo molecule generation, linker design, and binding affinity prediction), including the problem setup, representative methods, datasets, and evaluation metrics could act as a qualitative and a non-local metaphor for quantum homeopathy and as based on quantum theory could be also resulting to more novel fuzzy sphere formations ((Iconics1-4), (Eqs1-400), Supplementary Material METHODS AND MATERIALS)), (Figures1-99) and Quantum Deep Geometry generated drug designing processes providing explanations of the success of quantum homeopathic therapeutic approaches and an equivocal evidence for the efficacy of homeopathy [18-140] in quantum entanglement in general. [2-23,72-144] This is an advantage within this quantum homeo-formalism, because differential operators are formally ill-defined [14-148], although recent developments have shown that good results can still be achieved [26-159] also for other macroentanglement formation project. [14-149] These macro entanglement formations for Quantum Homeopathy based chemical geometrics would be far more useful for drug designers and researchers when trying to explore generalized entanglement as a vehicle for geometry processes. [6-88,89-150] In this situation, where quantum similarities between famous double-slit experiment of quantum physics, and quantum information processing are proposed docking free energy expectation values proving this apparent relationship between quantum homeopathic efficacy and QFT to QM reductions which could be explained in general terms of information loss from small molecule quantum superposition states. [34-98,94-158] The next step was to incorporate geometrical consciousness into this Quantum Homeopathy Model (Maths1-12,13a,13b,13c,13d,13e,13f,13g,13h,13i,13j,13k), ((Schematic1.(I-VI) (Supplementary Material METHODS AND MATERIALS)) (Eqs.1-400), ((Iconics1-4), (Eqs1-400), Supplementary Material METHODS AND MATERIALS)), (Supplement Material FUNCTIONS.1-25),
(Maths14a,14b,14c,14d,14e,14f,14g,15,16,17,18,19a,19b,19c,19d,19e,19f,19g,19h,20a,20b,20c,20d,20e,20f1,20f2,20f3,20g,20h,20i,20j,21a,21b,21c,21d,21e,21f,21g,21h,21i,21j,22a,22b,22c,22d,,22e,,22f,22g),(Cluster of BIOGENEA_ CONSENSUS_Eqs.1-26), and ((Figures 1- 133), (OUTPUTs1-3)), Supplementary Material METHODS AND MATERIALS)), (Ic1a), (Ic1b), (Ic1c), (Ic2a), (Ic2b), (Ic2c), (Ic2d,e,f,g,h,g,k,l,m,o,p,q,r), (Diagramm1, Diagramm2, Diagramm3), (Maths1-19) (Ic3a), (Ic3b) (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), (Ic3j),after translating this intimate relationship between water memory and protein folding issues, between drug substances in ultra-low concentrations and the quantum geometrics which is also a complementary one [21-99,101-159] when generating hypergeometric druggable fuzzy spheres (Figures1-100), ((Iconics1-4), (Eqs1-400), and Schematic1.(I-VI) Supplementary Material METHODS AND MATERIALS)) interacted with negative binding free energies accordingly. These functions can be deciphered through DockThor docking energy evaluations of the hydrophobic interactions among neighbored sidechains of an unfolded thermodynamic metastable state with great accuracy of the prediction. [1-195] Therefore, Ι suggest that the Practical consequences of this computational entanglement model for this quantum homeopathic research for mathematical chemistry practice could be more extended to new Generalized Entanglement Theoretical Drug Designing Models for understanding the effects of complementary and alternative medicine including Nano-and molecular events, thermodynamics/entropy, Quantum mechanisms and genetic instructions with the following inputs: ACE2 and AT1R receptors, Remdesivir’s therapeutic mechanism for the COVID-19, Candesartan’s role in ameliorating COVID-19 cytokine storm including Telmisartan’s molecular mechanism as tentative angiotensin receptor blocker and therapeutic agent for COVID-19. [28-137,150-189] To be made practical, these theoretical quantum algorithms are expected to require both significant refinement and effort in translation in the near term, since these refinements could include (i) recasting them for WOLFRAM devices that use the VQA, QAOA or QA frameworks for (ii) integrating greater biological context of the generalized entangled state (a) depends on the proper production process and is intimately connected to the ritual of remedy producing. [28-137,150-169] (OUTPUTs1-3), (Ic1a), (Ic1b), (Ic1c), (Ic2b), (Ic2c), (Ic2a), (Ic3a), (Ic3b) (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), (Ic3j), (Iconics4-6), and ((Iconics1-6), (Iconics1-4), (Eqs1-400), Supplementary Material METHODS AND MATERIALS)) Therefore, I hope that this work will stimulate the development of new directions in the study of the AT1R drug universe and other drug designing efforts. [40-162] Although it is less conclusive due to a large finite-size effect, we show that certain entanglement phases seem to exist even when a random state has permutation or translation symmetry for a non-vanishing boundary solution in a five-dimensional CS supergravity Quantum Foam [20-163] for all the poly-tetrahedron shaped pharmacophoric ligand provided here in the form of a Turing Machine Ruled Quantum Function which can carry U (1) charge among the unknown characteristics revealed in the cluster of the Gissitorviffirna_TM, Roccustyrna_gs, Roccustyrna_fr, and Roccustyrna_consv Quantum-computational structures. These AI-Quantum Homeopathy Entropy Negativities (QHEN) generated the highest total free energy values of negative docking energy factors and will be generalized into Spheroidal Wave Equations for Avogadro’s Number SphericalHarmonicY, ChebyshevT, and LegendreP Fuzzy Sphere-like small molecules, (Ic1a), (Ic1b), (Ic1c), (Ic3a), (Ic3b) (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), (Ic3j), (Ic2a), (Ic2b), (Ic2c) geometrical descriptors and (Eqs.1-400) ((Iconics1-4), (Eqs1-400), Supplementary Material METHODS AND MATERIALS)), which are ordinary differential equations (Eqs1-325), ((Iconics1-4), (Eqs1-400), and Schematic1.(I-VI) Supplementary Material METHODS AND MATERIALS)) with two regular singular points and one confluently irregular singular point. By incorporating Quantum Biological Evolution and QuantumTuring Deep Learning processes into this (Ic0a), (Ic0b), (Ic0c), (Ic1a), (Ic1b), (Ic1c), (Ic1d), (Ic2a), (Ic2b), (Ic2c), (Ic3a1), (Ic3a2), (Ic3b1), (Ic3b2), (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), and (Ic3j) Quantum Homeopathy Framework, this quantum homeopath of negative docking energy theory paradigm implying that this emergence of complexity with life which is an inherent feature of learning occurs throughout the history of the universe. With respect to these Quantum Homeopathy logic advantages, this led us to the Turing Machine Ruled Generalizations of Genetic, Homeopathic, and Clinical Data to Chemical Proteomic based Graph Translations (Ic3a), (Ic3c) as inputs into (Ic2e,f,g,h,g,k,l,m,o,p,q,r,q) these Quantum Circuit Generative Models of various number of qubits and quantum circuit layers discussed from above can yield operational docking energy advantages such as sample complexity advantages in particular which could have a great impact in future drug designing fields for the computer-aided designing of Hidden Druggable Pharmacophoric Subgroups and Small Molecule Triangularizations (Highlights Supplementary Material
Maths14a,14b,14c,14d,14e,14f,14g,19a,19b,19c, 19d,19e,19f,19g,19h,20a,20b,20c,20d,20e,20f1,20f2,20f3,20g,20h,20i,20j,21a,21b,21c,21d,21e,21f,21g,21h,21i,21j,22a,22b,22c,22d,,22e,,22f,22g), and (Cluster of BIOGENEA_ CONSENSUS_Eqs.1-26), (Diagramm1, Diagramm2, Diagramm3), (Highlights Supplementary Material, Maths1-21), and (Figures 1- 133), (Ic2e,f,g,h,g,k,l,m,o,p,q,r), ((Iconics1-4), (Eqs1-400), Supplementary Material METHODS AND MATERIALS)). With respect to these Quantum Homeopathy logic advantages, further experimental work is necessary to assess whether these diagonal-matrix potential advantages in variational (Ic2e,f,g,h,g,k,l,m,o,p,q,r) Quantum Circuit Generative Models of various number of qubits and quantum circuit layers for the Roccustyrna peptidemimetic Small Molecule Drug Discoveries discussed from above can yield operational docking energy advantages such as sample complexity advantages in particular which could have a great impact in future drug designing fields. These novel applications in the field of structure-based binding affinity prediction will have to address the points of criticism directed toward existing methods, therefore I will let your imaginations roam far outside the M87 supermassive rotating black hole in this structure-based molecular modeling section since only Avogadro’s Quantum Uncertainty relations between Entangled Space-Time backgrounds and Euclid Special Meta-Logic-Black Hole-Chemical Spaces (Highlights Supplementary Material,Maths19a,19b,19c,19d,19e,19f,19g,19h,20a,20b,20c,20d,20e,20f1,20f2,20f3,20g,
20h,20i,20j,21a,21b,21c,21d,21e,21f,21g,21h, 21i,21j,22a,22b,22c,22d,,22e,,22f,22g), and (Figures 1- 133), (OUTPUTs1-3)), (Ic1a), (Ic1b), (Ic1c), (Ic2b), (Ic2c), (Ic2a), (Ic3a), (Ic3b) (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), (Ic3j), ((Iconics4-6), (Supplementary Material METHODS AND MATERIALS)) for which the full Schwarzschild metric is required could be able of translating rare entropy signatures from Homeopathy Substances into 3D macromolecular structure representations for rational drug design, emphasizing to the most recent developments in both predictive and generative deep-learning methods.
Limitations of the Study
In this study, many amino acid features were utilized. Whether they are all actually beneficial for the prediction needs further exploration. In addition, all the information learned by these Quantum Deep Machine Learning approaches and Protein-ligand binding affinity prediction with edge awareness and supervised attention comes from the static crystal structure. Integrating the dynamic behavior information of protein–ligand complex in the model structure is still not well solved. Finally, more datasets are needed to verify the generalization ability and interpretability of these Edge Graph convolution-based and Supervised Attention-based Drug-Target Affinity prediction methodologies, where the super edge graph convolution can comprehensively be combined to Geometric Deep Learning for Structure-based Drug Design modules. Intuitively, sample complexity and generalization error as Quantum Hormetic advantages may arise when quantum entanglement enables the modeling of classically intractable correlative structures even if such sample complexity advantages are achievable with classical data, which will likely be problem instance specific [2-195], highly dependent on the distribution of the input data [1-99,2-195] and are unlikely to be superpolynomial [2-11,3-195]. However, a diverse and growing body of evidence from recent and previous work on novel and more efficient error correcting codes is required [1-14,1-16], for the realization of dynamical topological phases [34,4-35] and for the early demonstrations of logical qubits [31–43] by utilizing node and edge information and multi-supervised attention to efficiently learn the attention distribution that are consistent with real protein-ligand interactions proposing general conditions for the emergence of Turing patterns in a domain that changes size through homogeneous growth/shrinkage based on the qualitative changes of a potential function. Based on the Kolmogorov complexity of real weights, evolving weights, and real probabilities, respectively an infinite hierarchy of Bell pair Qubits of analog networks defined in terms of the Kolmogorov complexity should be Quantum Mechanically distributed at each molecular location of the Telmisartan’s chemical bonding that causes a semi Quantum negative downregulation effect inside the AT1 receptor at the mRNA and protein level apparently due to its action as a partial PPAR-gamma (Peroxisome Proliferator-Activated Receptor gamma) agonist. Moreover, while in the case of a Quantum Hormetic Regulator the limit of the continuum is reached (but not always) when the number of sites is huge and the spacing approaches zero, in our case there is the classic limit that is reached when the fluctuations of the quantum metrics vanish near the attractor. Moreover, while in the case of this Quantum Homeopathy Regulator the reduction of QFT to QM is not mathematically explicit, in this case it is, since the ansatz corresponds to the execution of a boson translation (as it was illustrated in Sections 1-3). Depending on the precise form, polynomial quantum speedups could be associated with useful quantum advantage, as even a polynomial classical algorithm does not mean that solutions can be obtained in a practical time. Both aspects may prove important in the further development of quantum algorithms in quantum chemistry. Other barriers to quantum advantages include (i) the sophistication of existing classical heuristic algorithms and the inherent parallelism of many of the problems they solve in a polynomial time computable noise p.s.d. for which the computation of its capacity cannot be performed in polynomial time, i.e., the number of computational steps on a Turing Machine grows faster than all polynomials (ii) the scale of both existing classical hardware and practical problem instances within the context of contemporary [3-185] ,function described by a polynomial time classical Turing machine, which is public; the client would like to sample a random x as the function input and use a protocol to send f(x) to the server (iii) the broad institutional support and incumbent advantage benefiting existing classical approaches (including extensive clinical validation in the medical setting) by introducing the Counter Turing Test (CT^2), a benchmark consisting of techniques aiming to offer a comprehensive evaluation of the robustness of existing AGTD techniques, and (iv) the likely precondition of FTQC to realize polynomial advantages based on amplitude amplification in practice revealing that synthetic data that passed visual Turing tests, can also enhance the representation learning capability of VisionFM, and leading to substantial performance gains on downstream ophthalmic AI tasks [41-190]. Thus, while current research in this direction shows long-term promise and should be explored further, many of these quantum advantages appear unlikely to be practical in the near term, including Turing-complete programming elements, programming languages, and sophisticated document notations. On the contrary, regarding the potential biological usefulness of entanglement as a communication system [30], it would be strange if biological systems had not used it showing that this landscape of quantum advantages considers the benefits of quantum computing technologies relative to existing classical alternatives by indicating that these geometric topology-driven heuristic algorithms used in this project are capable of fragmenting and remerging small molecules that could not be fragmented by the algorithm of any of the known reference databases. In fact, this holomorphic twist is equivalent to holomorphic BF-like Quantum fields theory as said before, since the computational skeletons of these innovative drug designs that are tetrahedrically quantized can finally assign definite values to the negative docking energy observables represented by noncommutative operators in this von Neumann’s scheme. Though still a controversial assertion, Quantum Homeopathy macroentanglement as a significant operational quantum advantage in the energy required to perform computations [4-27,4-36] gives much reason for optimism if the analysis offered here is at all useful and has some truth to it we have to suppose that the same Polynomial-time algorithms for prime factorization and discrete logarithms on a quantum computer process could play an important role in other therapeutic procedures as well. It is to be suspected that this meta-analysis of homeopathy supplies the quantum entanglement base of what is termed the quantum therapeutic alliance or was called rapport by the early hypnotists. All that happens in the meanwhile is unknowable, and the only theory I can make concerns the correlations among input and output events. [61-73] Different physical theories (for example, SU (2) vs. SL (2, r) Chern-Simons theory) correspond to quantizing different atoms that are hidden in QFT just like Quantum information does (remember that there is an uncertainty relationship between Quantum Geometrics and Quantum information) considering these CS equations (Highlights Supplementary Material,
Maths19a,19b,19c,19d,19e,19f,19g,19h,20a,20b,20c,20d,20e,20f1,20f2,20f3,20g,20h,20i,20j,21a,21b,21c,21d,21e,21f,21g,21h,21i,21j,22a,22b,22c,22d,,22e,,22f,22g), (Eqs1-325), ((Iconics1-4), (Eqs1-400), and Schematic1.(I-VI) Supplementary Material METHODS AND MATERIALS)) as relative CS motion equations of nearby atoms, and a coordinate-independent measure of the wave’s effects described by the geodesic deviation function. I claim that a quantum computer would be needed in order to simulate the hidden quantum network (HQN) of the designed quantum circuits under this study. But using a binary tree to trace the original QFT would mean not always recovering the Quantum Characteristics of the latter in four dimensions, whose field content consists of (?k) ((Supplement Material) FUNCTION1-28). For this reason, in particular, a regulator would be needed to play the role of a hidden quantum definitional function [53-72] to allow the switch from a classical metalanguage to a hypergeometric language logic in this quantum homeopathy version [43-73] of a definitional function that allows to pass from a QML to the quantum logic of quantum information (QLI). [39-73] More precisely, a 100 qubit quantum computer is required in order to be programmed to be in a one-to-one correspondence with the HQN as had already pointed out in these Quantum Functions and QFT Loss Quantum Function (QLQF) established here as a direct connection between processes of Quantum Chemical Biology and Turing Machine Learning Ruled Euclid Special Spaces which are consisted of a connected part, the molecular interaction of this disconnected part that calculates in parallel the total free energy fields. From a mathematical perspective, these quantum circuits could also recover Hilbert Symmetric Spaces as may be called by Chern–Simon’s theory. Thus, the problem is in QP, the quantum analogue of the class P, and if we wish, more realistically, to model the oracle's size and running time, then we could assume the oracle size, in general f, to be O(N), this being the size of the oracle which simply contains a ROM list of the function values. With these results, I expect to take advantage of the knowledge accumulated in half a century of classical networking research and operation to create strong Quantum Homeopathy Network Architectures and guide the experimental focus for the development of Quantum Negative Energy Repeaters for the implementation of real-world Quantum based Drug Design protocols. Implementation of these in-silico Quantum Homeopathic Phase Cryptographic Experiments led me to Quantum Turing Machine Networking Acceleration and fragmentations of the existed molecular networks into NPs occupying electronic orbitals that are involved in the endothelial damage of the cardiovascular system in an early stage of the COVID-19 disease as reflected by the release of highly sensitive troponin and natriuretic peptides (NPs). Notably, while Quantum Turing may yield superpolynomial Quantum Homeopathy, advantages on classically hard problems, whether these near-term algorithms can fully capitalize on the computational power afforded by quantum information remains a matter of theoretical investigation (e.g. [4-124]). Despite some advancement, [3-79,135-149], these Quantum Mechanics and Quantum-gravitational ideas provided us along only with a Theoretical Quantum Gravity background from Quantum Homeopathy Information into a Quantum Homeopath Folding Translation Scheme (Ics.3a,3b,3c) which is highly dependent on the folding of typical secondary structures as the means to hierarchically pave a negative docking energy and native folding pathway and still exhibit limitations in terms of prediction accuracy and require significant efforts in feature design. This research has highlighted the application of geometric deep learning methods, which represent the protein-ligand complex structure as 3D grids or 3D graphs for processing and predicting these Bell Pair Qubits at another chemical space solution if the intersect would reduce these negative docking energies to a finite set of Haag’s theorems. [1-48] Since not all computational groups working in this field will have the expertise, equipment, or desire to perform the required synthesis and experimental testing, collaborations with experimentalists will be highly valuable. To comprehensively evaluate the utility of emerging new models in a real-world drug design context, experimental validation of the proposed molecular structures is paramount. Still, we believe that quantum computing remains a potentially powerful model of computation. Quantum computers can quickly solve some problems not known complete such as factoring and the potential to solve problems such as graph isomorphism Highlights.
SupplementaryMaterial,Maths112,13a,13b,13c,13d,13e,13f,13g,13h,13i,13j,13k,19a,19b,19c,19d,19e,19f,19g,19h,20a,20b,20c,20d,20e,20f1,20f2,20f3,20g,20h,20i,20j,21a,21b,21c,21d,21e,21f,21g,21h,21i,21j,22a,22b,22c,22d,,22e,,22f,22g), ((Iconics1-4), (Eqs1-400), and Schematic1.(I-VI) Supplementary Material METHODS AND MATERIALS)), (Ic0a), (Ic0b), (Ic0c), (Ic1a), (Ic1b), (Ic1c), (Ic1d), (Ic2a), (Ic2b), (Ic2c), (Ic3a1), (Ic3a2), (Ic3b1), (Ic3b2), (Ic3c), (Ic3d), (Ic3d), (Ic3e), (Ic3f), (Ic3g), (Ic3h), (Ic3i), and (Ic3j) for finding a short vector in a lattice. Moreover, quantum computing can give a large increase in speed, for example a quadratic improvement in NP-like search problems.
Declaration
The paper reports annotated quantum functions, and cluster of equations regarding Avogadro Number’s oriented HyperGeometric and ChebyshevT Functions on Black Hole Paradox Generalizations and Turing Machine Ruled Quantum Homeopathy Water Memory Entanglements for the Translation of COVID19 Homeopathy Remedies into the Neprilysin and ACE2/AT1R receptors targeted DRVYIHPFX- ligands. This study generated new and unique drug designs. This project contains the following extended data: https://zenodo.org/records/10149911
Ethical approval and consent to participate.
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Ethical Guidelines
Not applicable.
Data and Software Availability statement
The paper reports annotated quantum functions, and cluster of equations regarding Avogadro Number’s oriented HyperGeometric and ChebyshevT Functions on Black Hole Paradox Generalizations and Turing Machine Ruled Quantum Homeopathy Water Memory Entanglements for the Translation of COVID19 Homeopathy Remedies into the Neprilysin and ACE2/AT1R receptors targeted DRVYIHPFX- ligands. This study did generate new and unique drug designs. This project contains the following extended data: https://zenodo.org/records/10149911.
Funding
No funding received for this research article.
Competing interest
No potential competing interest was reported by the author.
Author’s Contribution
Grigoriadis Ioannis's diverse contributions to the published work are accurate and agreed. Grigoriadis Ioannis wrote the whole manuscript and solely contributed to multiple roles:
- Conceptualization, Ideas, formulation, or evolution of overarching research goals and aims.
- Methodology, Development, or design of methodology; creation of models.
- Writing-Review & Editing, Preparation, creation, and presentation of the published work by those from the original research group, specifically critical review, commentary, or revision including pre-or post-publication stages.
- Visualization, Preparation, creation, and presentation of the published work, specifically visualization/data presentation.
- Supervision, Oversight, and leadership responsibility for research activity planning and execution, including mentorship external to the core team.
- Project administration, Management, and coordination responsibility for research activity planning, and execution.
Acknowledgements
I would like to deeply express my special thanks of gratitude to my teacher (George Grigoriadis Pharmacist) as well as My CEO and principal (NikolAoΨοs Grigoriadis Phd Pharmacist) who gave me the golden opportunity to do this wonderful project on Grover Search Quantum Deep Learning Chemistry topics, which also helped me in doing a lot of Original drug Repurposing and drug Combination Research and I came to know about so many new things I am thankful to them.
- Abstract
- Keywords
- Summary
- Introduction
- Methods & Materials
- Results
- Discussions
- Concluding Remarks
- Limitations of the Study
- Declaration
- Consent for publication
- Competing interests
- Ethical Guidelines
- Data and Software Availability statement
- Funding
- Competing interest
- Author’s Contribution
- Acknowledgements