Extracellular Matrix Graft with PHMB Versus High Osmolarity Surfactant
Matthew Regulski1, Garth James2, Erika Avera3, Matthew F. Myntti4*
- The Wound Institute of Ocean County, NJ, 54 Bey Lea Road Tom’s River, NJ 08759, USA.
- Montana State University Center for Biofilm Engineering, Bozeman, MT, 366 Barnard Hall Bozeman, MT 99717, USA.
- Montana State University Center for Biofilm Engineering, Bozeman, MT, 366 Barnard Hall Bozeman, MT 99717, USA.
- Next Science™ LLC, 10550 Deerwood Park Blvd, Suite 300, Jacksonville, FL 32256, USA.
*Corresponding Author: Matthew F. Myntti, Next Science™ LLC, 10550 Deerwood Park Blvd, Suite 300, Jacksonville, FL 32256, USA, 124001, TEL: 855-564-2762 ; FAX: 855-564-2762; E-mail:mmyntti@nextscience.org
Citation: Matthew Regulski, Garth James, Erika Avera, Matthew F. Myntti (2018) Extracellular Matrix Graft with PHMB Versus High Osmolarity Surfactant J Dermatol & Ther 2:112.
Copyright:© 2018 Matthew F. Myntti, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received date: December 11, 2018; Accepted date: December 20, 2018; Published date: December 24, 2018.
Abstract
Background: According to the National Institutes of Health, biofilms are associated with 80% of persistent microbial infections within the body; but biofilms can also form virtually anywhere, including on and under expensive graft materials. Additionally, the bacteria within biofilms are highly resistant to many conventional therapies. The purpose of this study is to evaluate and compare the prevention of biofilm growth on an antimicrobial wound matrix versus a biofilm-disrupting technology (a high osmolarity surfactant), using the Montana State University Center for Biofilm Engineering colony drip-flow reactor (cDFR).
Methods: The cDFR models in vitro biofilm growth, mimicking the chronic wound environment. The biofilms were formed from methicillin-resistant Staphylococcus aureus strain 10943 and Pseudomonas aeruginosa strain 215, chronic wound clinical isolates from Southwest Regional Wound Care Center in Lubbock, TX. These strains were inoculated, applied unto a polycarbonate membrane, dried, and covered with gauze, the antimicrobial wound matrix, and the biofilm-disrupting gel. One membrane served as a control with nothing applied. Exudate, via flow of growth medium, was introduced at 5ml/hr per channel and the cDFR was incubated at 33°C for 24 hours. Three repeat experiments were conducted and mean CFU/membrane and Log Reduction (LR) were calculated.
Results: Plate count results indicated significant reductions for P. aeruginosa (over 8 log) and MRSA (over 6.8 log) in biofilms treated with the high osmolarity surfactant. Reductions for the antimicrobial wound matrix material were lower and not significantly different from the gauze. Confocal scanning laser microscopy supported plate count results, indicating live bacteria within the gauze, mixture of live and dead bacteria with the antimicrobial wound matrix, and mostly dead bacteria from the biofilm-disrupting technology.
Conclusions: The use of biofilm-disrupting technology on graft materials should be more effective in biofilm growth prevention. Furthermore, biofilm-disrupting technology has wide-reaching implications for treating persistent infections and removing barriers to wound healing.
Keywords:
Biofilm, chronic wounds, graft materials
Introduction
All wounds, especially chronic ones, become colonized with bacteria. It arises from the patient's skin, respiratory tract, gastrointestinal tract, or where exogenous bacteria are transferred from the environment or conveyed on the hands of healthcare workers [1-3]. When the bacterial load reaches 105 CFU/G, which is colony forming units per gram of tissue, the wound is considered infected; when levels are greater than or equal to 106 CFU/G or when greater or equal to four species are present, failure to heal may occur [4,5]. The formation of microbial biofilms within wounds has now been associated with a failure to heal [6-8]. Compared with their planktonic counterparts, microorganisms within a biofilm are highly resistant to environmental stresses and host immune responses and are tolerant to antimicrobials [9-12]. Most of these infections begin with colonization by Gram-positive cocci, followed by different genera of Gram-negative bacilli from the host or the environment; over time the infections become polymicrobial, consisting of numerous species including obligate anaerobes [13-16]. Once formed, the biofilm prevents wound contracture and epithelization, dysregulates the host immune response by affecting intracellular signaling, and damages the tissue through a combination of chronic inflammation and microbial virulence factors [17-19].
In nature, as well as at different infection sites, microorganisms can exist within a protective structure termed biofilm, which consist of mushroom-like multicellular structures that are surrounded by an extracellular polymeric substance or extracellular polysaccharide matrix (EPS) formed from bacterial and host products [20,21]. Biofilm infected wounds are difficult to treat. Encasement of the bacterial pathogens within the EPS, combined with the presence of necrosis from the surrounding wound bed and decreased blood flow to the compromised areas, prevents host defense responses from reaching the infected tissue and eliminating the infection and protects the pathogens within from the systemically administered antibiotics [22-24]. Strategies to treat wound biofilms are multi-faceted, including debridement to remove the biofilm and necrotic tissue, wound dressings to control moisture in the wound bed and to protect granulating tissue from damage, and treatment of the wound bed with topical antimicrobials to prevent recurrence of the microorganism [25-27]. Bacterial biofilms start to form when planktonic bacterial cells adhere to the wound surface by attaching to the exposed extracellular matrix proteins. Within about two hours, the bacteria rapidly begin expressing extracellular polymeric substances and up to 800 new proteins to form a microcolony [28]. Medially secreted structures intrinsically block the host immune cells, common antibiotics, and biocides from reaching the bacterial cells; thus, different treatment strategies than those used for planktonic bacteria are required in such cases [29]. Therefore, the objective of this study was to test and to compare the prevention of biofilm growth on an antimicrobial wound matrix (PuraPly; Organogenesis; Canton, MA) and in a biofilm disrupting technology (BlastX; Next Science; Jacksonville, FL) used to protect graft material, using the Montana State University CBE colony drip-flow reactor (cDFR). Untreated gauze was also tested for comparison.
Methods and Materials
The cDFR is a model for growing in vitro biofilms that mimic the chronic wound environment and has been used for both single species [30,31] and polymicrobial biofilms [32]. It utilizes the standard drip-flow biofilm reactor (DFR 100-4, Biosurface Technologies Corp., Bozeman, MT) with an absorbent pad and membranes. The membranes are inoculated and fresh, flowing nutrients are supplied. The nutrients are wicked upwards through the pad, feeding the microorganisms on the membrane, and mimicking wound exudate. The biofilm forms on the top of the membrane. The 2.5 cm diameter absorbent pads (AP1002500, Merck Millipore Ltd, Cork IRL) were attached to the centers of glass microscope slides (48300-047, VWR International) using a small drop of silicone adhesive and the slides were placed in the reactor. The reactor was then sterilized by autoclave. Immediately prior to inoculation, the absorbent pads were hydrated with the growth medium, 10%-strength brain-heart infusion broth (Difco™ 237200, Becton Dickenson, Sparks, MD) and ultraviolet-sterilized 13 mm diameter 0.22 µm pore size polycarbonate membranes (GE Water & Process Technologies, Trevose, PA) were placed on the absorbent pads.
Mixed-species biofilms were formed using methicillin-resistant Staphylococcus aureus (MRSA) strain 10943 and Pseudomonas aeruginosa strain 215. These are chronic wound clinical isolates obtained from the Southwest Regional Wound Care Center in Lubbock, TX and are maintained at -70°C as a frozen stock cultures in the CBE. Inocula were grown overnight from frozen stock cultures in 10%-strength brain-heart infusion broth (BHI, Difco™ 237200, Becton Dickenson, Sparks, MD). The P. aeruginosa culture was diluted 1:10 with phosphate-buffered saline (PBS) and then mixed 1:1 with the MRSA culture and 10 µL of the mixture was applied to the center of the polycarbonate membrane. The inoculum was dried for 15 minutes and then the gauze, PuraPly, and BlastX were applied. One membrane had nothing applied, to serve as an untreated control. Flow of growth medium (exudate) was then initiated at a rate of 5 ml/hr per channel and the CDFR was incubated at 33°C (approximate wound temperature) for 24 hours.
For plate counts, each sample was placed in 10ml of sterile double-strength D/E Neutralizing Broth (Becton, Dickinson, and Company, Sparks, MD) to neutralize the treatments. The samples were then subjected to 30 seconds of vortexing and two minutes of sonication, followed by an additional 30 seconds of vortexing. Serial 10-fold dilutions were made using sterile PBS, and the dilutions were plated on Pseudomonas Isolation Agar and Staphylococcus Medium 110 (Becton, Dickinson, and Company, Sparks, MD). After 24 hours of incubation at 37°C, the plates were counted and the colony forming units (CFU) per membrane was calculated. Three repeat experiments were conducted and mean CFU/membrane and Log Reduction (LR) were calculated. The LR for PuraPly and BlastX were compared by ANOVA using Minitab software and a 95% confidence interval.
For confocal scanning laser microscopy (CSLM), the dressing/membrane pairs were removed from the reactor and placed on a microscope slide, and saturated with LIVE/DEAD BacLight Bacterial Viability Kit (Life Technologies, Carlsbad, CA). The sample was incubated in the dark for 10 minutes at room temperature. The samples were then covered with 1.5% noble agar and examined using a Leica SP5 confocal scanning laser microscope. Image processing was done using Imaris™ software.
Results
The P. aeruginosa and S. aureus biofilm prevention plate count results are shown in Figure 1. For P. aeruginosa, CFU counts in all three experiments were below detection limits for BlastX-treated samples, which indicated reduction of over 8-log (mean 8.41 LR). This was significantly different from the gauze treated samples (p=0.010), and the PuraPly material (p=0.047). The PuraPly-treated samples had a more variable reduction (mean 3.31 LR) and were not significantly different from gauze (p=0.170).
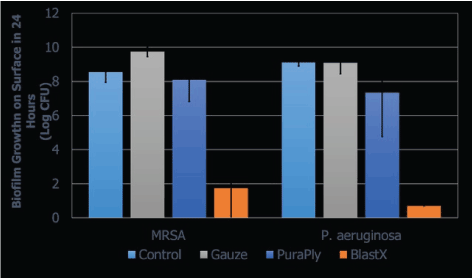
Figure 1: The mean growth of P. aeruginosa and S. aureus after 24 hours from the control; gauze; and PuraPly and BlastX treated samples shown in log CFU. Samples not under the horizontal bars are statistically significant (p-value of <0.05). The mean biofilm growth was calculated from 3 repeated experiments.
The BlastX treatment also had a high reduction of MRSA (mean 6.81 LR), which was significantly different from both the gauze (p=0.003) and PuraPly (p=0.008) treatments. The MRSA reduction for the PuraPly treatment (mean 0.45 LR) was not significantly different from gauze (p=0.350).
Overall, the CSLM results, as shown in Figure 2, agreed with the plate count results. Bacteria within the gauze-treated control biofilms emitted primarily green fluorescence indicating live bacteria. The PuraPly -treated biofilms emitted both red and green fluorescence, indicating a mixture of live and dead bacteria. In contrast, the BlastX-treated biofilms emitted primarily red fluorescence indicating that most of the bacteria were dead.
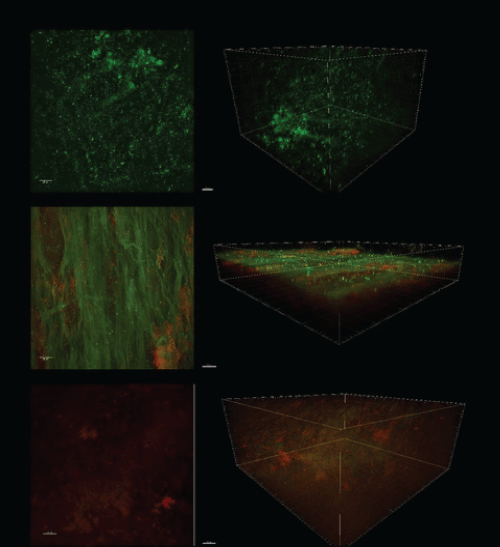
Figure 2: The 2D and 3D images showing the fluorescence of the stained living (green) and dead (red) bacteria taken using the confocal scanning microscope. Images are from the control and the PuraPly- and BlastX-treated biofilms.
Discussion
According to the National Institutes of Health (United States), over 80% of persistent microbial infections within the body involve biofilms. Prevalent examples of biofilm associated infections include chronic lung infections, periodontitis, endocarditis, and osteomyelitis [33-35]. The cost of infections represents a major portion of the healthcare budget, and these costs continue to grow in exponential rates [36].
Diabetic extremity ulcerations develop in approximately 20% of people with diabetes and are the leading cause of hospitalization and amputation among such patients. Wound infection, faulty wound healing, and ischemia in combination with foot ulcers are the most common precursors to diabetes related amputations. 85% of lower limb amputations in patients with diabetes are preceded by biofilm infected foot ulcerations [37-39]. More than 80,000 amputations are performed on the United States diabetic population each year [40]. Diabetic foot ulcer infections followed by amputations contribute dramatically not only to the morbidity among persons with diabetes, but are also associated with severe clinical depression and dramatically increased mortality rates [41]. Such infected ulcers resulting in amputation account for a three-fold increase risk of death within 18 months. As such, diabetic foot ulcers are the most common, disabling, and costly complications of diabetes [42,43].
A primary impediment to the healing of chronic wounds is biofilm phenotype infections [44-45]. Biofilms, by definition, are the ubiquitous and natural phenotype of bacteria. They typically consist of the polymicrobial populations of cells, which are attached to a surface and encase themselves in the hydrated extracellular polymeric substances. Microbial populations that have attached to biological or nonbiological surfaces are the most basic description of medical biofilm. Thus, most chronic infections, including bacterial, that are associated with chronic wounds, do exist in biofilm [46-48]. Thus, bacteria that reside within these mature biofilms are highly resistant to many traditional therapies. New strategies must be developed so that we can treat these biofilms effectively, safely, and expeditiously. The longer these wounds stay open, the greater the risk and increase in morbidity and mortality.
In this study, we compared antimicrobial wound matrix PuraPly, which is touted as having biofilm-destroying capabilities through the incorporation of polyhexamethylene biguanide (PHMB). This antimicrobial agent is an antiseptic. It is a synthetic compound that has a chemical structure similar to antimicrobial peptides (AMPs) that occur naturally in keratinocytes and neutrophils. Naturally occurring AMPs are produced as a normal immune response and have antibacterial, antiviral, and antifungal affects [49,50].
The high osmolarity surfactant (BlastX) has the capability to bind to the metal ions within the EPS to be able to pull these polymers out into solution, in effect “unzipping” the biofilm and exposing the bacteria that lie deep within the biofilm. Bacteria within the EPS typically occupy 5% to 30% of the volume of the biofilm. The thickness or dimension of cell clusters in the biofilm can range from a few microns to a few millimeters. Nutrients and metabolic waste either diffuse directly through the biofilm or are transported through open water channels [51,52]. By utilizing BlastX, and through the high osmolarity and surfactant, we are able to disrupt and swell the dormant bacterial biofilm and are able to lyse not only the biofilm, but also any planktonic bacteria that are found on the surface of the biofilm. In biofilms, poor antibiotic penetration, nutrient limitation, slow growth, adaptive stress responses, and inclusion of phenotypic variants are shown to mediate resistance to antibiotics and to biocides [53]. In certain conditions, biofilm bacteria have a ten-fold higher intercellular survival rate than planktonic bacteria [54]. This multitude of defense mechanisms created by the biofilm can enhance the chronicity and persistence of chronic wounds, therefore increasing morbidity and mortality.
In this study, we show that the biofilm disrupting technology had a significantly large statistical degradation of biofilms produced by two very virulent and commonly encountered bacteria in chronic wounds, while the antimicrobial wound matrix performance was no better statistically than gauze in the treatment of these biofilms.
Therefore, this biofilm disrupting technology demonstrates significant degrading and destructive effects upon frequently encountered chronic wound bacterium and was far superior to the antimicrobial wound matrix. Biofilm disruption promises to remove one of the greatest barriers to healing and will substantially improve wound closure rates, outcomes, and cost savings.
Conclusions
The use of the antimicrobial matrix did not induce a statistically significant decrease in biofilm growth as compared to the gauze control. In comparison to the antimicrobial matrix material, the use of biofilm disrupting technology yielded a statistically significant 8-log reduction in Pseudomonas aeruginosa and a 6.8-log reduction in Methicillin-Resistant Staphylococcus aureus (MRSA) biofilm growth. As such, the use of biofilm disrupting technology on graft materials should be substantially more effective in preventing biofilm growth.
References
- Church D1, Elsayed S, Reid O, Winston B, Lindsay R (2006) Burn wound infections. Clin Microbiol Rev 19: 403-434. [crossref]
- Rafla K, Tredget EE (2011) Infection control in the burn unit. Burns 37: 5-15. [crossref]
- Gist S, Tio-Matos I, Falzgraf S, Cameron S, Beebe M (2009) Wound care in the geriatric client. Clin Interv Aging 4: 269-287. [crossref]
- Black CE, Costerton JW (2010) Current Concepts Regarding Infected Wound Microbial Ecology and Biofilms on Wound Healing. SURG. CLIN. North AM. 90: 1147-1160.
- Bowler PG, Duerden BI, Armstrong DG (2001) Wound microbiology and associated approaches to wound management. Clin Microbiol Rev 14: 244-269. [crossref]
- Percival SL, Emanuel C, Cutting KF, Williams DW (2012) Microbiology of the skin and the role of biofilms in infection. Int Wound J 9: 14-32. [crossref]
- Ricco JB, Thanh Phong L, Schneider F, Illuminati G, Belmonte R, et al. (2013) The diabetic foot: a review. J Cardiovasc Surg (Torino) 54: 755-762. [crossref]
- Percival SL, Hill KE, William DW, Hooper SJ, Thomas DW, et al. (2012) A Review of the Scientific Evidence for Biofilm in Wounds. Wound Repair Regen. 20: 647-657.
- Donlan RN, Costerton LW (2002) Biofilms Survival Mechanisms of Relevant Microorganisms. CLIN. Microbiol. Rev. 15: 157-193.
- Hřiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O (2010) Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents 35: 322-332. [crossref]
- Leid JG, Wilson CJ, Shirtliff ME, Hassett DJ, Parsek MR, et al. (2005) The Exopoly Alginate Process Pseudomonas Aeruginosa Biofilm Bacteria. ISN-Gamma-Mediated Necrophase Healing. J. Immunol. 175: 7512-7518.
- Schaber JA, Triffo WJ, Suh SJ, Oliver JW, Hastert MC, et al. (2007) Pseudomonas Aeruginosa Forms Biofilms in Acute Infection Independent of Cell-to-Cell Signaling. Infect. Immun. 75: 3715-3721.
- Kennedy P, Brammah S, Wills E (2010) Burns, biofilm and a new appraisal of burn wound sepsis. Burns 36: 49-56. [crossref]
- James GA, Swogger E, Wolcott R, Pulcini Ed, Secor P, et al. (2008) Biofilms in chronic wounds. Wound Repair Regen 16: 37-44. [crossref]
- Malic S, Hill KE, Hayes A, Percival SL, Thomas DW, et al. (2009) Detection and Identification of Specific Bacteria in Wound Biofilms Using Peptide Nucleic Acid Fluorescent in Situ Hybridization. (PNA FISH) Microbiol. 155: 2603-2611.
- Edwards R, Harding KG (2004) Bacteria and wound healing. Curr Opin Infect Dis 17: 91-96. [crossref]
- Hart J (2002) Inflammation. 2: Its role in the healing of chronic wounds. J Wound Care 11: 245-249. [crossref]
- Ovington L (2003) Bacterial toxins and wound healing. Ostomy Wound Manage 49: 8-12. [crossref]
- Posluszny JA Jr, Conrad P, Halerz M, Shankar R, Gamelli RL (2011) Surgical burn wound infections and their clinical implications. J Burn Care Res 32: 324-333. [crossref]
- Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284: 1318-1322. [crossref]
- Pruitt BA Jr, McManus AT, Kim SH, Goodwin CW (1998) Burn wound infections: current status. World J Surg 22: 135-145. [crossref]
- Shirani KZ, Vaughan GM, Mason AD Jr, Pruitt BA Jr (1996) Update on current therapeutic approaches in burns. Shock 5: 4-16. [crossref]
- Ammons MC (2010) Anti-biofilm strategies and the need for innovations in wound care. Recent Pat Antiinfect Drug Discov 5: 10-17. [crossref]
- Davis SC, Martinez L, Kirsner R (2006) The diabetic foot: the importance of biofilms and wound bed preparation. Curr Diab Rep 6: 439-445. [crossref]
- Wolcott RD, Kennedy JP, Dowd SE (2009) Regular debridement is the main tool for maintaining a healthy wound bed in most chronic wounds. J Wound Care 18: 54-56. [crossref]
- Bjarnsolt T, Kirketerp-Moller K, Kristiansen S, Phipps R, Neilson AK, et al. (2007) Silver Against Pseudomonas Aeruginosa Biofilms. APMIS 115: 921-928.
- Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DJ, et al. (2002) Pseudomonas Aeruginosa Displays Multiple Phenotypes During Development as a Biofilm. J. Bacteriol. 184: 1140-1154.
- Wolcott R, Dowd S (2011) The role of biofilms: are we hitting the right target? Plast Reconstr Surg 127 Suppl 1: 28S-35S. [crossref]
- Singh PK. Schaefer AL, Parsek MR, Moninger TO, Welch MG, Greenberg E, et al. (2000) Quorum-sensing Signals Indicate that Cystic Fibrosis Lungs are Infected with Bacteria Biofilms. Nature 407: 762-764.
- Agostinho AM, Hartman A, Lipp C, Parker AE, Stewart PS, et al. (2011) An in vitro model for the growth and analysis of chronic wound MRSA biofilms. J Appl Microbiol 111: 1275-1282. [crossref]
- Lipp C, Kirker K, Agostinho A, James G, Stewart P (2010) Testing wound dressings using an in vitro wound model. J Wound Care 19: 220-226.
- Woods J, Boegli L, Kirker KR, Agostinho AM, Durch AM, et al. (2012) Development and application of a polymicrobial, in vitro, wound biofilm model. J Appl Microbiol 112: 998-1006. [crossref]
- Distel JW, Hatton JF, Gillespie MJ (2002) Biofilm formation in medicated root canals. J Endod 28: 689-693. [crossref]
- Gristina AG, Oga M, Webb LX, Hobgood CD (1985) Adherent bacterial colonization in the pathogenesis of osteomyelitis. Science 228: 990-993. [crossref]
- Smith A (2004) “Etiology of the Problem Wound” in Wound Care Practice, edited by DJ Scheffield, CE Phipps, A Smith, p 3-48, Best Publishing Company, Flagstaff.
- Palumbo PJ, Melton I (1985) “Peripheral Vascular Disease in Diabetes” in Diabetes in America: Diabetes Data Compiled in 1984, Government Printing Office, Washington, D.C.
- Adler AI, Boyko EJ, Ahroni JH, Smith DG (1999) Lower Extremity Amputation in Diabetes. The Independent Facts of Peripheral Vascular Disease, Sensory Neuropathy, and Foot Ulcers. J. Diabetes Care 22:1029-1035.
- Pecoraro RE, Ahroni JH, Boyko EJ, Stensel VL (1991) Chronology and determinants of tissue repair in diabetic lower-extremity ulcers. Diabetes 40: 1305-1313. [crossref]
- National Diabetes Information Clearing House (2005) National Diabetes Statistics. HTPP://Diabetes.NIDKK, NIH, Gov/Pubs/Statistics/Index. HTM#7.
- Ismail K1, Winkley K, Stahl D, Chalder T, Edmonds M (2007) A cohort study of people with diabetes and their first foot ulcer: the role of depression on mortality. Diabetes Care 30: 1473-1479. [crossref]
- Ramsey SD, Newton K, Blough D, McCulloch K, Sandhu M, et al. (1999) Patient-Level Estimates of the Costs of Complications of Diabetes in a Managed-Care Population. Pharmacoeconomics 16: 285-295.
- Ramsey SD, Newton K, Blough D, McCulloch DK, Sandhu N, et al. (1999) Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care 22: 382-387. [crossref]
- Bjarnsholt T, Kirketerp-Mřller K, Jensen PŘ, Madsen KG, Phipps R, et al. (2008) Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen 16: 2-10. [crossref]
- James GA, Swogger E, Wolcott R, Pulcini Ed, Secor P, et al. (2008) Biofilms in chronic wounds. Wound Repair Regen 16: 37-44. [crossref]
- Wolcott RD, Rhoads DD (2008) A study of biofilm-based wound management in subjects with critical limb ischaemia. J Wound Care 17: 145-148, 150-152, 154-155. [crossref]
- Percival SL, Bowler P, Woods EJ (2008) Assessing the effect of an antimicrobial wound dressing on biofilms. Wound Repair Regen 16: 52-57. [crossref]
- Wolcott RD, Ehrlich GD (2008) Biofilms and chronic infections. JAMA 299: 2682-2684. [crossref]
- Dowd SE, Sun Y, Secor PR, Rhoads GD, Wolcott RE, et al. (2008) Survey of Bacterial Diversity in Chronic Wounds Using Pyrosequencing, DGGE, and Full Ribosome Shotgun Sequencing. BMC Microbiol 8: 43.
- Dowd SE, Zaragoza J, Rodriguez JR, Oliver MJ, Peyton RP (2005) Windows. NET Network Distributed Basic Local Alignment Search Toolkit. (W. ND-BLAST) BNC. Bioinformatics 6: 93.
- Compensate Document (2010) PHNB and its Potential Contribution to Wound Management. WUK: Aberdeen. (Level IV Evidence).
- Lindholm C (2010) Technology Update: Understanding the Role of PHMB: A Topical Approach to Wound Infection. Wounds Int.; (1) 3. (Level IV Evidence).
- Gray D, Barett S, Battacharyya M, Butcher M, Enoch S, et al. (2010) PHMB and Its Potential Contribution to Wound Management. Wounds UK; 6(2) 40-46 (Level IV Evidence).
- Stewart PS (2002) Mechanisms of antibiotic resistance in bacterial biofilms. Int J Med Microbiol 292: 107-113. [crossref]
- Spiliopoulou AI, Kolonitsiou F, Krevvata MI, Leontsindis M, Wilkinson TS, et al. (2012) Bacterial Adhesion, Intracellular Survival and Cytokine Induction Upon Stimulation of Mononuclear Cells with Planktonic or Biofilm Phase Staphylococcus Epidermidis. FENS Microbiol. LEPT: 330-356.