Germ-Line Mutanome Profiling of The Breast Cancers in Pakistani Population
Sadia Ajaz1*, Sani-e-Zehra Zaidi1, Saleema Mehboob Ali1, Aisha Siddiqa2, Muhammad Ali Memon2
1 Dr. Panjwani Center for Molecular Medicine and Drug Research (PCMD), International Center for Chemical and Biological Sciences (ICCBS), University of Karachi, Karachi, Pakistan.
2 Atomic Energy Medical Centre (AEMC), Jinnah Postgraduate Medical Centre (JPMC), Karachi, Pakistan.
*Corresponding Author: Sadia Ajaz, Assistant Professor, Molecular Oncology (P-035) Lab, Dr. Panjwani Center for Molecular Medicine and Drug Research (PCMD), International Center for Chemical and Biological Sciences (ICCBS), University of Karachi, Karachi, Pakistan, TEL:+922134824924-5 ; FAX:+922134819018-9 ;E-mail:sadiaajaz.pcmd@iccs.edu
Citation: Sadia Ajaz, Sani-e-Zehra Zaidi, Saleema Mehboob Ali, Aisha Siddiqa, Muhammad Ali Memon, et al. (2018) Germ-Line Mutanome Profiling of The Breast Cancers in Pakistani Population. Arch Mol Med & Gen 1:106.
Copyright: : © 2018 Sadia Ajaz, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received date: October 24, 2018; Accepted date: November 05, 2018; Published date: November 08, 2018.
Abstract
Statement of the Problem: Cancers are complex disorders. Consanguineous populations, by virtue of reduced genetic diversity, provide a unique model for the investigations of the underlying genetic component(s). A prototype example in the breast cancers is the identification of BRCA1/2 gene defects in Ashkenazi Jews. In Pakistan, the age-standardized rate (ASR) of breast cancer incidence in females is among the highest in Asia, whereas the morality rate is one of the highest in the world. With the consanguinity rate of 56.4% and inbreeding coefficient (F) of 0.0331, it is extremely important to investigate the role of inherited mutations in breast cancers in Pakistani population.
Methodology and Theoretical Orientation: BROCA analysis for breast cancers consists of twenty-seven (27) established and candidate breast cancer genes involved in molecular carcinogenesis. A pilot hospital-based cohort study was designed. Eighty-five breast cancer patients and three controls with no medical history of any cancer, participated in the study. The BROCA investigations were carried out by a genomic capture, massively parallel next generation sequencing assay on Illumina HiSeq2000 assay with 100bp read lengths. Copy number variations were determined by partially-mapped read algorithm. Once the mutation was identified, it was validated by Sanger sequencing. After informed consent, the mutations were screened in the familial samples.
Findings: The analysis revealed germ-line mutations in 12% of the patients. These mutations were restricted to three genes (BRCA 1, BRCA 2, and TP53). The identified mutations consist of both novel and previously reported alterations and result in protein truncation. No mutations were identified in the remaining twenty-four (24) genes. Mutation screening in the familial samples identified carriers in four out of five families.
Conclusions and Significance: The study provides framework for the development of preventive and treatment strategies against breast cancers in Pakistani population.
Keywords
Breast cancer, Susceptibility, Candidate genes, Consanguinity, Pakistani population.
Introduction
It is estimated that approximately 20-25% cases of breast cancers arise due to highly penetrant hereditary component. The information is mainly influenced by data from Caucasian populations. Pakistani population is highly consanguineous. The consanguinity rate is 56.4% [1] and inbreeding co-efficient is 0.0331 (Hussain and Bittles, 1998). Such populations provide a unique model to investigate and quantify the role of candidate genes and/or specific mutations in complex disorders. The hallmark example is the discovery of BRCA1&2 mutations in breast cancer patients among Ashkenazi Jews [2]. Information on genetic basis of breast cancers in Pakistani population is scarce. The region has one of the highest age-standardized incidence rates (ASIR) of breast cancers in Asia. Breast cancer-related mortality rates are amongst the highest in the world [3]. Despite the disease burden, little is known about the underlying factors, genetic and/or environmental, for breast cancers in this population.
Since the discovery of its role in breast cancer susceptibility (Hall et al., 1990) screening of BRCA1 has been carried out in different populations around the globe. In addition, a number of genes encoding proteins involved in double strand DNA break repair, mismatch DNA repair, cell cycle, cell adhesion, transcription, DNA synthesis have been implicated in inherited predisposition to breast cancers (BROCA)
The present pilot study is the first-systematic attempt to investigate the role of all known breast cancer genes in Pakistani population. We report the investigation of germline mutations in twenty-seven breast cancer candidate genes in a cohort of breast cancer patients from Pakistan. The study has significant implications in delineating the molecular pathology of inherited breast cancers.
Methods
Ethics Statement
The project is in accordance with the Declaration of Helsinki - 2013 [4]. Informed consent was obtained from the participants. The project was approved by Ethical Review Committees of collaborating institutions: Atomic Energy Medical Centre (AEMC), Jinnah Postgraduate Medical Centre (JPMC), and Dr. Panjwani Center for Molecular Medicine and Drug Research (PCMD), International Center for Chemical and Biological Sciences (ICCBS), University of Karachi, Karachi, Pakistan.
Sampling
The pilot study, conducted from July 2016 to June 2017, included blood samples from ninety-four (94) breast cancer patients and two (02) healthy volunteers. 8-10ml of blood samples were transferred to the ACD-coated vacutainers (BectonDickinson®, Franklin Lakes, NJ, USA). In case of healthy volunteers, 5ml blood samples were collected. The samples were either processed immediately or stored at 4°C until DNA isolation. After quality and quantity control, eighty-five (85) samples were analysed by next generation sequencing (NGS).
Cases and Family History Instrumentation
Patients volunteered the information about the family history of breast cancer, or any other cancer, number of pregnancies, number of children, the age of menarche and age of menopause. Details on Bloom Richardson grading system and tumor node metastasis (TNM) staging as recommended by the Union International Contre le Cancer (UICC) were obtained from the hospital medical records with the patient’s consent.
DNA Extraction, Quantification, and Quality Control
Standard Phenol-Chloroform method was used for the extraction of DNA from the WBCs of patient’s blood sample [5]. Isolated DNA was quantified on UV spectrophotometer (Beckman Coulter® DU 530). The acceptable range of 260/280 ratios was set as 1.8-1.99. Quality control was carried out on 0.7% agarose gel and visualized under UV light using Azure C-300® imaging system. None of the extracted DNA samples showed the shredded or streaked pattern on the agarose gel (Figure 1).
Genomic Capture, Massively Parallel Next Generation Sequencing Assay (BROCA Analysis)
After DNA extraction, quantity and quality control, germline DNA was sequenced using breast and ovarian cancer gene panel (BROCA). This assay sequences all the exonic and flanking intronic sequences of the selected genes. The analysis is able to capture all types of mutations including large genomic re-arrangements in known breast cancer genes [6-8]. The breast cancer gene panel assay investigates following 27 genes: BRCA1, BRCA2, TP53, ATR, BARD1, BRIP1, FAM175A, FANCM, GEN1, MRE11A, NBN, RAD51B, RAD51C, RAD51D, RECQL, RINT1, SLX4, BAP1, PALB2, PTEN, STK11, XRCC2, ATM, CHEK1, CHEK2, CDH1, and CTNNA1.
Exon Amplification and Sanger Sequencing
The exons in BRCA1, BRCA2, and TP53 genes carrying mutations as identified by N.G.S. were amplified. The amplicons were purified by an isopropanol-based method. 15µl of the purified amplified products along with the primers were used for sequencing. Sanger sequencing of samples was performed commercially.
Bioinformatic Analysis
ExAC and NHGRI databases were used as reference for the identification of novel mutation(s).
Results and Discussion
10 samples from 85 patients (approx.12%) had a germ-line mutation. These were confined to BRCA1, BRCA2, and TP53 genes.
BRCA1
In total five mutations were identified in BRCA1 in case series of 85 patients. Among these two are novel. Details of a representative mutation are provided below:
c. 68_69del (Exon 2) Mutation
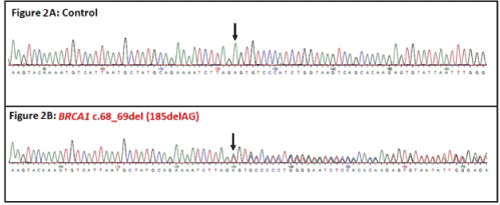
The mutation started at 17: 41276045 (hg37). It was validated by Sanger sequencing (Figure 2A and 2B). The consequence of mutation is 23frameshift leading to stop codon at amino acid position 39 and a truncated protein.
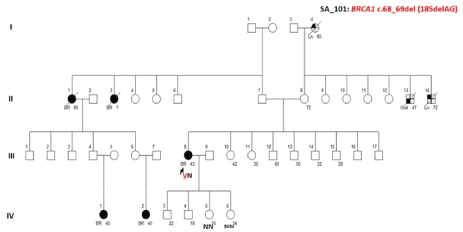
Age of the Proband and Familial Segregation Analysis
Proband is a 43 years old female. After the parental consent, c.68_69del germline mutation analysis in DNA samples from daughters of the patient revealed the absence of mutation (Figure 3).
Age of the Proband and Familial Segregation Analysis
Proband is a 43 years old female. After the parental consent, c.68_69del germline mutation analysis in DNA samples from daughters of the patient revealed the absence of mutation (Figure 3).
Comparison of Identified Mutation with Published Reports from Different Populations
The mutation along with two other founder mutations (BRCA1 5382insC and BRCA2 6174delT) accounts for 60% of hereditary cases of breast cancers in Ashkenazi Jews [9]. It contributes to 16%-20% of breast cancer cases diagnosed before the age of 50 years in this population. The mutation has 2068 BIC database entries. It has been identified in American, Ashkenazi Jewish, Ashkenazi/Central European, Austrian, Belgian, British, Canadian, Central/Eastern Europe, Chilean, Czech, Dutch, French, German, Hispanic, Indian, Italian, Latvian, Malaysian, Mexican American, Native American, Norwegian, Spanish, USA, Western Europe, Western Europe/Ashkenazi populations (NCBI).
This is the sixth report of c.68_69 (185delAG) mutation from Pakistan (6/686: 0.87%) and the first report of this mutation in an Urdu-Speaking ethnic group. The mutation has previously been identified in patients belonging to Punjabi and Pathan ethnicity. The geographical location of reported mutations is shown in the map of Pakistan (Figure 4).
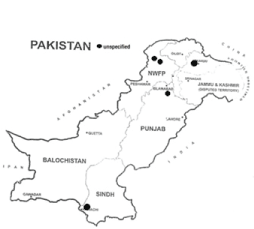
Figure 4: Region-wise distribution of c.68_69del (185delAG) mutation reported from Pakistan.
Discussion
BRCA1 mutations play a significant role in genetic predisposition to cancers. It consists of 24 exons. The mutations are more frequent in exons encoding three specific domains of BRCA1 protein (Figure 5). The first is RING (Really Interesting New Gene) domain encoded by exons 2-7. The second domain is encoded by exons 11-13, which comprises 65% of the BRCA1 protein sequence. It includes two Nuclear Localization Signals (NLS), and a serine-rich component. It is also the region where key DNA repair proteins, RAD50 and RAD51, as well as c-Myc (a transcription factor) and Retinoblastoma (a cell cycle regulator) binds. The third domain is BRCA1 C-terminal (BRCT) domain. It recognizes pSer-X-X-Phe sequence and forms a complex with its phosphorylated binding partners. These include BACH1, CtIP, and CCDC98/abraxas [10].
The RING domain of the BRCA1 plays a key role in its association with BARD1 (Figure 6A), with consequent ubiquitin ligase activity and nuclear retention of two proteins. c.68_69del (185delAG) results in Glu23fs and stop codon at position 39. The transcript does not undergo non-sense mediated mRNA decay (NMD) and results in a truncated protein (Figure 6B). It is known to affect apoptotic pathway, chemosensitivity, and transcription/gene regulation. In specific populations, it is the most frequently reported founder mutation [11].
The RING domain of the BRCA1 plays a key role in its association with BARD1 (Figure 6A), with consequent ubiquitin ligase activity and nuclear retention of two proteins. c.68_69del (185delAG) results in Glu23fs and stop codon at position 39. The transcript does not undergo non-sense mediated mRNA decay (NMD) and results in a truncated protein (Figure 6B). It is known to affect apoptotic pathway, chemosensitivity, and transcription/gene regulation. In specific populations, it is the most frequently reported founder mutation [11].
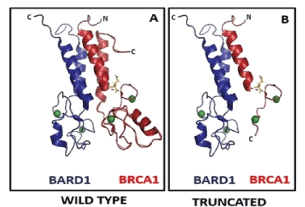
The present study reports c.68_69 as candidate BRCA1 germline founder mutation, underlying predisposition to breast cancers in the local population. Despite the limited sample size in previous and present reports, this germline mutation has been identified in two patients from Punjabi ethnicity, two from Pathan ethnicity, and one where ethnicity is unknown. The present study reports this mutation in a patient belonging to Urdu-speaking ethnicity, with a strong family history of multiple primary tumors including five cases of breast cancers. These recurrent reports indicate that the mutation is a strong candidate founder mutation in this population.
Taking into account the present study as well as previous reports [12,13], the frequency of this mutation in breast cancer patients from Pakistani population is close to that of Ashkenazi Jews. It is possible that the mutation may have arisen prior to separation of the Jewish diaspora, about 2000 years ago [14]. There is also some evidence that the mutation may have arisen twice, once in Jewish and once in non-Jewish population. However, the data is inconclusive. It has been proposed, that the reason for its persistence in different populations is possibly as yet unidentified biological advantage [15]. The evolutionary insight into the mutation is as yet an intriguing conundrum, which might hold the potential in translational medicine.
In conclusion, the present study provides evidence for a founder mutation in Pakistani population. The determination of recurrent mutation frequency has important implications in devising strategies for genetic testing, counselling, and treatment of familial/hereditary breast cancers in the indigenous population.
BRCA2
In total three BRCA2 mutations were identified in a case series of 85 patients. Details of a representative mutation are provided below:
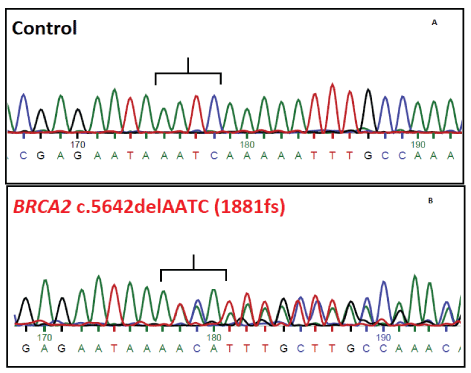
c.5642delAATC (Exon 11) Mutation
The mutation started at 13: 32914134 (hg37). It was validated by Sanger sequencing. The consequence of mutation is 1881frameshift leading to stop codon at amino acid position 1891 and a truncated protein (Figure 7).
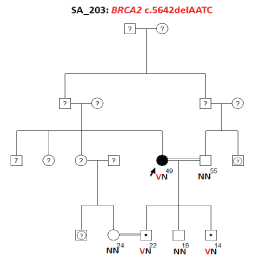
Age of the Proband and Familial Segregation Analysis
The age of the patient is 49 years. After informed consent, c.5642delAATC (exon 11) mutation was investigated in three sons, a niece, and husband (paternal cousin) of the proband. Two of the sons, aged 14 and 22, were identified as carriers of the germline mutation. Figure 8.
Analysis of Germline Mutations
Comparison with NHGRI database shows that although 1881fs has been reported earlier, underlying germline mutation c.5642delAATC is being reported for the first time.
Discussion
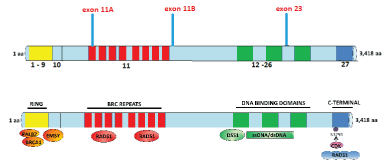
Exon 11 of BRCA2 encodes domain with conserved BRC repeats (Figure 9). It is through this domain that BRCA2 forms a complex with Rad51. Thus, through interaction with this recombinase, the role of BRCA2 in double strand break (DSB) repair was established. The formation of BRCA2-Rad51 complex involves two domains of BRC repeats, the other one at the C-terminus [6]. The binding at BRCA2 C-terminus leads to oligomerization of Rad51. During homologous DNA repair (HDR), DSBs are first cleaved and converted into single stranded breaks (ssDNA) coated by Rad51 nucleoprotein filaments, which facilitate the invasion of ssDNA into the homologous sister strand. The invading ssDNA serves as a primer for DNA synthesis, using the undamaged sister strand as the template, enabling the strand exchange. Thus, HDR is error-free [16]. Mutation in BRCA2 exon 11, is therefore likely to result in the accumulation of DSBs and genome instability.
TP53
In all, two mutations in TP53 gene were identified in a case series of 85 patients. Details of a representative mutation are provided below:
W91X (Exon 4) Mutation
The mutation started at 17: 74579415 (hg37). It was validated by Sanger sequencing. The consequence of mutation is protein truncation at amino acid position 91 (Figure 10).
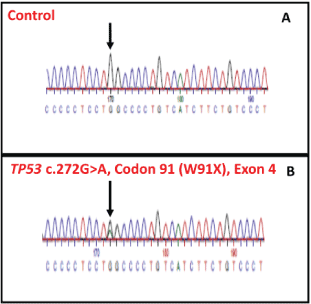
Age of the Proband and Familial Segregation Analysis
The age of the patient is 49 years. Pedigree and different cancers identified in the family are shown in figure 11.
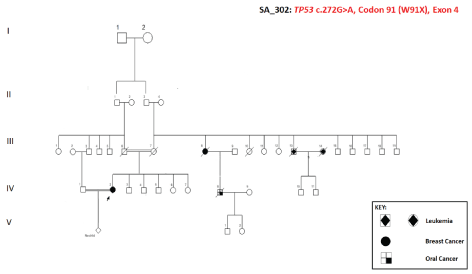
Analysis of Germline Mutations. Comparison with reference databases showed that this is the first time W91X germline TP53 mutation is reported from any population.
Discussion:
TP53 is known to be the guardian of genome [17]. It plays a central role in multiple primary cancers. Although a high number of somatic mutations have been reported, the germline mutations are comparatively rare [18]. As evidenced by BRCA1 and BRCA2 genes in Ashkenazi Jews, consanguineous populations provide a unique model for the identification and quantification of role of specific genetic alterations in cancer pathology. A previous study from different region of the country investigated 105 familial/early onset cases of breast cancer for germline mutations in TP53. However, they did not find any nonsense mutation in their series [19]. The differences in population sub-stratification and the methodology must be taken into consideration while comparing these two studies. In the present study the investigations were carried out by a genomic capture, next generation sequencing assay with 100bp read lengths (BROCA).
W91X mutation arising from G>A substitution in exon 4 was identified in a 48-year-old Gujrati patient, who reported strong family history of breast cancers. This is the first time this germline mutation is being reported. Recent data suggests that TP53 truncating mutations are likely to have neomorphic potential.
Interestingly, at least some of the truncated p53 mutants are able to evade nonsense-mediated decay, resulting in promotion of metastasis and tumour manintainance similar to gain-of-function mutants [20]. Figure 12.
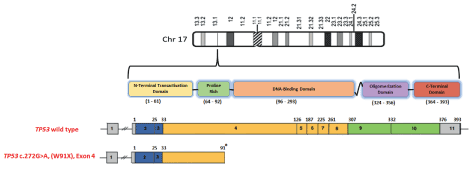
Given the emerging data regarding the functional role(s) of truncated p53 and present report of one such mutations, TP53 may be considered in familial cases of breast cancers [21-29]. The present study has shown that genetic component other than BRCA1 and BRCA2 plays a definitive role in susceptibility to breast cancers in the local population [30-37].
In conclusion, the present report is the first comprehensive study of twenty-seven breast cancer genes in representative cases from Pakistan [37-42]. The discovery study shows that mutations in one of three genes, BRCA1, BRCA2, and TP53 comprise the germline mutanome profile of breast cancers in Pakistani population [43-48].
Additional Information
Authors’ contributions: SA-Study design, Sampling, Benchwork, Analysis, and Manuscript Preparation; SZZ - Benchwork, Analysis; SMA- Sampling, Benchwork, Analysis; AS – Sampling, Medical Data Collection, MAM - Sampling, Medical Data Collection;
Acknowledgments
The research was funded in part by the ICCBS recurrent grant. Prof. MC King and Dr. Tom Walsh (Department of Genome Sciences and Medical Genetics, University of Washington, Seattle, Washington, USA) were instrumental in BROCA analysis and its sponsoring. The staff at AEMC, JPMC, Karachi for their facilitation, and transport office at the ICCBS for sampling. Finally, we are especially grateful to the patients and their families who participated in the study.
Conflicts of Interest
The authors declare no potential conflicts of interest.
Financial Statement
The funders had no role in study design, data collection, analysis, and decision to publish or preparation of the manuscript.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2013 [4]. Informed consent was obtained from all patients and participants included in the stud
References
- Bittles AH, Black ML (2015) Global patterns & tables of consanguinity. URL http://consang. net.
- Lord CJ, Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16: 110-120. [crossref]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, et al. (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer 136.
- World Medical Association Declaration of Helsinki (2013): Ethical Principles for Medical Research Involving Human Subjects. JAMA. 310: 2191-2194.
- Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. third. Cold Spring Harbor Laboratory Press, New York.
- Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, et al. (2010) Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proceedings of the National Academy of Sciences 107: pp.12629-12633.
- Nord AS, Lee M, King MC, Walsh T (2011) Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics 12: 184. [crossref]
- Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, et al. (2011) Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proceedings of the National Academy of Sciences 108: pp.18032-18037.
- Walsh T, Mandell JB, Norquist BM, Casadei S, Gulsuner S, et al. (2017) Genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi Jewish women. JAMA oncology 3: pp.1647-1653.
- Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D, et al. (2012) Structure-function of the tumor suppressor BRCA1. Computational and structural biotechnology journal 1: pp.1-8.
- Buisson M, Anczuków O, Zetoune AB, Ware MD, Mazoyer S, et al. (2006) The 185delAG mutation (c. 68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Human mutation 27: pp.1024-1029.
- Liede A, Malik IA, Aziz Z, Rios Pd Pde L, Kwan E, et al. (2002) Contribution of BRCA1 and BRCA2 mutations to breast and ovarian cancer in Pakistan. Am J Hum Genet 71: 595-606. [crossref]
- Rashid MU, Naeemi H, Muhammad N, Loya A, Yusuf MA, et al. (2016) A novel deleterious c. 2656G> T MSH2 germline mutation in a Pakistani family with a phenotypic overlap of hereditary breast and ovarian cancer and Lynch syndrome. Hereditary cancer in clinical practice 14: p.14.
- Bruchim Bar-Sade R, Kruglikova A, Modan B, Gak E, Hirsh-Yechezkel G, et al. (1998) The 185delAG BRCA1 mutation originated before the dispersion of Jews in the diaspora and is not limited to Ashkenazim. Human molecular genetics 7: pp.801-805.
- Laitman Y, Feng BJ, Zamir IM, Weitzel JN, Duncan P, et al. (2013) Haplotype analysis of the 185delAG BRCA1 mutation in ethnically diverse populations. European Journal of Human Genetics 21: pp.212-216.
- Lee H (2014) Cycling with BRCA2 from DNA repair to mitosis. Exp Cell Res 329: 78-84. [crossref]
- Malkin D, et al. (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233-1238.
- Blanco A, Grańa B, Fachal L, Santamarińa M, Cameselle-Teijeiro J, et al. (2010) Beyond BRCA1 and BRCA2 wild-type breast and/or ovarian cancer families: germline mutations in TP53 and PTEN. Clin Genet 77: 193-196. [crossref]
- Rashid MU, Muhammad N, Amin A, Loya A, Hamann U, et al. (2017) Contribution of brca1 large genomic rearrangements to early-onset and familial breast/ovarian cancer in Pakistan. Breast Cancer Res. Treat. 161: 191-201.
- Shirole NH, et al. (2016) Tp53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife. 5.
- Ahmad J, Calvez-Kelm L, Daud S, Voegele C, Vallee M, et al. (2012) Detection of BRCA1/2 mutations in breast cancer patients from Thailand and Pakistan. Clinical genetics 82: pp.594-598.
- Alvarez C, Tapia T, Perez-Moreno E, Gajardo-Meneses P, Ruiz C, et al. (2017) BRCA1 and BRCA2 founder mutations account for 78% of germline carriers among hereditary breast cancer families in Chile. Oncotarget 8: p.74233.
- Breast Cancer Information Core (BIC) database: https://research.nhgri.nih.gov/projects/bic/Member/index.shtml
- Drayna D (2005) Founder mutations. Sci Am 293: 78-85. [crossref]
- Karczewski KJ, Weisburd B, Thomas B, et al. (2017) The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 45: D840-840D845. [crossref]
- Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, et al. (2007) Founder mutations in BRCA1 and BRCA2 genes. Annals of Oncology 18: pp.vi93-vi98.
- Genecards®. Human Gene Database: http://www.genecards.org/cgi-bin/carddisp.pl?gene=BRCA1 (last accessed: 30th December 2017).
- Górski B, Jakubowska A, Huzarski T, Byrski T, Gronwald J, et al. (2004) A high proportion of founder BRCA1 mutations in Polish breast cancer families. International journal of cancer 110: pp.683-686.
- Krześniak M, Butkiewicz D, Rachtan J, Matuszczyk I, Grzybowska E, et al. (2017) A novel germline TP53 mutation p.Pro190Arg detected in a patient with lung and bilateral breast cancers. Adv Med Sci 62: 207-210. [crossref]
- Jauhri M, et al. (2017) Prevalence and coexistence of kras, braf, pik3ca, nras, tp53, and apc mutations in Indian colorectal cancer patients: Next-generation sequencing-based cohort study. Tumour Biol 39: 1010428317692265.
- Kastenhuber ER, Lowe SW (2017) Putting p53 in Context. Cell 170: 1062-1078. [crossref]
- Levy-Lahad E, Catane R, Eisenberg S, Kaufman B, Hornreich G, et al. (1997) Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. American journal of human genetics 60: p.1059.
- Li FP, Fraumeni JF Jr. (1969) Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 71: 747-752.
- Li FP, Fraumeni JF Jr (1982) Prospective study of a family cancer syndrome. JAMA 247: 2692-2694. [crossref]
- Moatter T, Aban M, Khan S, Azam I, Pervez S, et al. (2011) BRCA1 status in Pakistani breast cancer patients with moderate family history. Journal of the College of Physicians and Surgeons Pakistan 21: p.680.
- Nakatsuka N, Moorjani P, Rai N, Sarkar B, Tandon A, et al. (2017) The promise of discovering population-specific disease-associated genes in South Asia. Nature genetics 49: p.1403.
- National Center for Biotechnology Information: https://www.ncbi.nlm.nih.gov/
- Hsiao TH, Chiu YC, Hsu PY, et al. (2016) Differential network analysis reveals the genome-wide landscape of estrogen receptor modulation in hormonal cancers. Sci Rep 6: 23035. [crossref]
- Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008. [crossref]
- Orban TI, Olah E (2003) Emerging roles of BRCA1 alternative splicing. Molecular Pathology 56: p.191.
- PED 6.0.2 – Pedigree Drawing Software: http://www.medgen.de/
- Petrucelli N, Daly MB, Pal T (1993) Brca1- and brca2-associated hereditary breast and ovarian cancer in GeneReviews® (ed. Adam, M.P.) (University of Washington, Seattle, 1993 – 2017).
- PyMol: https://pymol.org/2/
- Rashid MU, Zaidi A, Torres D, Sultan F, Benner A, et al. (2006) Prevalence of BRCA1 and BRCA2 mutations in Pakistani breast and ovarian cancer patients. International journal of cancer 119: pp.2832-2839.
- Romeo G, Bittles AH (2014) Foreward. Consanguinity and genomics. Hum Hered 77: 5. [crossref]
- Roy R, Chun J, Powell SN (2011) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12: 68-78. [crossref]
- Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, et al. (2014) p53Ψ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci U S A 111: E3287-3296. [crossref]
- Thorlacius S, Sigurdsson S, Bjarnadottir H, Olafsdottir G, Jonasson JG, et al. (1997) Study of a single BRCA2 mutation with high carrier frequency in a small population. American journal of human genetics 60: p.1079.