Theoretical Structural Model of The Glycolytic Enzyme Phosphoglycerate Kinase C from Leishmania Mexicana Mexicana: Transition from A Continuous to Discontinuous Helix at the C-Terminus and The Role of GXXXG Motif
Sashank Srinivasan1, Sangita Aggarwal2, Vidya Raghunathan3*
1 Former research scholar, National Institute of Immunology, New Delhi, India.
2 ARSD College, Delhi University, New Delhi, India.
3 National Institute of Immunology, New Delhi, India.
*Corresponding Author: Vidya Raghunathan, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi, India, 110067, TEL: +919810213191 ; FAX: +911126703776;E-mail:vraghuna@nii.ac.in
Citation: Sashank Srinivasan, Sangita Aggarwal, Vidya Raghunathan (2018) Theoretical Structural Model of The Glycolytic Enzyme Phosphoglycerate Kinase C from Leishmania Mexicana Mexicana: Transition from A Continuous to Discontinuous Helix at the C-Terminus and The Role of GXXXG Motif. Allergy drugs clin immunol 2:109.
Copyright:© 2018 Vidya Raghunathan, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received date: August 04, 2018; Accepted date: August 13, 2018; Published date: August 16, 2018
Abstract
Phosphoglycerate kinase C (PGKC) from the trypanosomatid, Leishmania is an important housekeeping enzyme whose 63 residue C-terminal extension is believed to be important in glycosome compartmentalisation. Structural studies with this enzyme have been experimentally challenging. We sought to establish the three dimensional structure of residues 6-417 of PGKC_Lmexicana by homology modelling and biochemical data. Since PGKC of Leishmania and T. brucei are evolutionarily related and have high sequence homology the known structure of the PGKC_Tbrucei was used as template. We have created a .pdb library of individual fragments of the enzyme based on the Jpred secondary structure which can be useful for experts in the field of bioinformatics/protein modelling. The final theoretical 3dimensional model of L.mexicana PGKC (residues 1-479) enables visualization of the GXXXG motif in the enzyme fold. Our biochemical data and the docking interactions reveal new aspects of the tertiary fold of PGKC_Lmexicana. Fresh conjectures on PGKC’s role in the glycosome, give crucial leads for further research.
Keywords
Phosphoglycerate kinase C; Leishmania; glycosomal membrane; model structure; GXXXG motif; transport.
Introduction
3-Phosphoglycerate kinase (EC 2.7.2.3; PGK) is a metabolic enzyme of the glycolytic pathway which synthesizes ATP (Adenosine triphosphate) by transfer of a phosphate group from 1,3-bisphosphoglycerate (1,3–BPG) to adenosine dinucleotide phosphate (ADP), its globally known function in all organisms. However auxiliary functions have been seen in specific instances [1-3]. For example, PGK of Group B streptococcus (GBS) is expressed on the cell surface where it binds actin and plasminogen through four lysine residues and effects bacterial virulence [2]. In Sendai virus transcription, PGK is involved in interaction with tubulin and forming an initiation complex that promotes mRNA synthesis [1]. Similarly, in Bamboo mosaic virus chloroplast PGK is recruited to target the viral complex into the chloroplast [3]. In each of these cases the enzymatic function of PGK that is, its traditional role in the glycolytic pathway, is not required for the newly observed function. These examples highlight a membrane association potential for PGK. PGK represents a shining case of multifunctionality arising from overcoming evolutionary challenges at both the structural level as well as in folding.
In trypanosomatids, PGK is a metabolic enzyme as in all other organisms, but in addition, is a metabolic regulator of the glycolytic flux [4]. An important feature of glycolysis in kinetoplastida is the presence of glycosomal and cytoplasmic isoforms of the glycolytic enzymes. PGK exists in 2 or 3 isoforms in kinetoplastida. The first half of glycolysis is compartmentalized within the glycosome, PGK being at the bifurcation of glycolytic pathway into the cytoplasm. In Leishmania only two isoforms of PGK are found. The cytoplasmic PGK is represented as PGKB and the glycosomal isoform is PGKC. Until recently, when size specific metabolite pores were found in the glycosomal membrane, [5], the glycosome was thought to be completely impermeable. Specific transporter and antiporters where considered to be involved in transport of glycolytic intermediates from glycosome to the cytoplasm. The metabolite pores provide alternate kinetic models of glycosomal function which explain the control of glycolysis and transport through the glycosomal membrane without involvement of antiporters [5]. According to the work of Achcar et al., [6] these metabolite pores do not support transport of ADP or ATP. Compartmental segregation of PGK in Leishmania will certainly be useful in controlling ATP/ADP ratios especially during metabolic stress.
The only known structure of trypanosomatid phosphoglycerate kinase is that from T. brucei (PGK_Tbrucei), ), the causative agent of African trypanosomiasis [7]. No Leishmania PGK structures are known so far. PGK is a classic example of a hinge-bending enzyme; a monomer of two almost equal-sized domains separated by a well-structured inter-domain region. Many crystal structures of PGK show an open and closed conformation, the former in which the two domains, called N-domain and C-domain are well separated, and the later in which the two domains come closer by a large rigid body angular movement (of N-domain with respect to the C-domain) of about 7.7 degrees. The C-domain is where the nucleotide substrate (ADP or ATP) binds and the N-domain binds to the triose substrate (1,3- BPG or 3phosophoglycerate (3-PG)) [8]. The hinge-bending hypothesis forms the basis of the catalytic activity of PGK. The 9 or so available structures of PGK confirm that large conformational changes follows the binding of the two substrates. Structure of the PGK_Tbrucei was the first closed conformation observed [7]. Studies comparing the free enzyme structure with that of binary complexes with substrates shows that the nucleotide binding site is partially buried and that nucleotide binding does not cause any conformational change. The triose phosphate binding site involves positively charged residues like guanidinium group of 3 arginines and a histidine residue. The carboxyl group of 3-PG interacts with a water molecular that is H-bonded to the triple glycine at alpha helix 14 and is also H-bonded to Arg 39 in helix 1. The changes in the protein conformation upon binding both substrates are known by comparing the X-ray structures of the binary and ternary complexes of PGK with substrates, which are now described. The entire alpha helix 14 undergoes a large piston like shift with respect to the N-domain upon binding of both substrates [7]. Further, enhanced interactions of the carboxylate group of 3-PG with Arg 39 and Arg 39 with Thr 393 (alpha helix 13, C-domain) lead to domain closure and active catalytic conformation. Another important residue K219 at the C-domain moves about 10Å closer to the 3-PG binding site on the N-domain in the ternary complex. G376 (alpha helix 13) and G399 (alpha helix 14) form interactions with the phosphate groups of 3-PG.
The hinge region between the domains is made of alpha helix 13, which is part of the Cdomain, strand βL and α-helix 14 which folds into the N-domain. While βL forms the main molecular hinge of PGK, α-helix 7 is also part of the hinge. Many inter-domain residue interactions involving electrostatic, hydrophobic as well as hydrogen bonding are seen in the open conformation. A very large inter-domain hydrophobic cluster interaction involving phenylalanine, alanine, histidine, valine and leucine has been noted [7]. A fully closed conformation was obtained in the crystals structure of hPGK (human PGK1) further confirming these observations [9].
The glycosomal isoform, PGKC from Leishmania is the largest PGKC known with 479 residues and molecular weight of 51 kD. The cytoplasmic isoform, PGKB is 417 residues in length like yeast, mammalian or porcine PGK’s. The 63 residue C-terminal extension is unique to PGKC’s of genus Leishmania. It is considered that this extension is important for compartmentalization based on the observation in Trypanosoma congolense that both PGK isoforms are cytosolic in absence of the C-terminal extension. It has been often stated that the Leishmania PGKC’s does not contain the peroxisomal targeting signal (PTS-1 or PTS-2) used by other proteins for glycosomal localization. However there is a -TKL tripeptide just two residues prior to the end of the protein at position 475 – 477 very similar to a PTS-1, which is –SKL. In a recent proteomic profiling of the Leishmania donovani glycosome, Jamdhade et al. [10], analysed thousands of proteins for PTS-1 and PTS-2 and found that PTS-1 may exist in conserved variants or also appear in unusual PTS-1 motifs. The highly variable nature of PTS-1 motifs in kinetoplastida has been noticed earlier [11-15]; PTS-1 may be strong or weak and have auxiliary motifs immediately upstream of them [10]. One such auxiliary motif may be presence of basic residues, 10 residues upstream. Leishmania PGKC has the PTS-1 and the upstream basic residues in its 63-mer region. Since PGKC_Lmexicana interaction with peroxin 5 (PEX5) receptor protein is not established we cannot with certainty say right now what the function of this PTS-1 dodecamer signal is in the protein.
We have previously shown that the 63 residue extension of PGKC_Lmexicana has a transmembrane helix starting from residue 451 ending with double arginine residues at positions 471 and 472 [16]. We also highlighted the presence of a GXXXG motif which has been shown in literature to be a key motif important for transmembrane dimerization of proteins bearing the motif within the membrane a helix [17,18]. Here we present the theoretical model of PGKC_Lmexicana to get a more detailed insight into the structure of this region of the protein. The 63-mer extension of PGKC_Lmexicana being a large domain without any prior structural data available, could not be incorporated into any model of PGKC_Lmexicana by homology modelling or docking. We therefore used PGKB_Lmexicana as a template in biochemical interaction studies with peptides derived from 63-mer extension. From this knowledge base a full model of PGKC_Lmexicana could be made, validated and analysed. A prominent hydrophobic patch on the surface of PGKC_Lmexicana is found containing residues which from part of the transmembrane helix. The GXXXG motif may have an important role in in stabilization of the enzyme fold.
Materials and Methods
Protein–Peptide interaction
The three peptides, 11-mer, 18-mer and 25-mer (Table 1) used in the study were custom synthesized by BioSynthesis Incorporated, Texas, USA. All reagents used in the assays and binding studies were purchased from Sigma Chemical Co., USA. Recombinant PGKB_Lmexicana was expressed in E. coli and purified as described earlier [16]. All peptides were tested for solubility and their circular dichroism spectra recorded in water, methanol and trifluoroethanol. 11-mer and 18-mer were soluble in aqueous buffer. Whereas 11-mer was mostly unstructured, the 18-mer had an extended conformation. 25-mer was insoluble in aqueous buffer, its circular dichroism showed a complete transition to a helical conformation in trifluoroethanol and methanol solvents. In the presence of PGKB_Lmexicana all three peptides solubilized. Different molar ratio solutions of PGKB and peptides were initially used during standardization experiments ranging from 1:0.1 PGKB:peptide to 1:4 PGKB:peptide. 12.5 uM stock solution of PGKB_Lmexicana and 100 uM peptide stock solution in 50 mM Tris, pH 7.6 were used. For comparison between peptides a ratio of 1:1 PGKB:peptide was chosen; 6.25μM protein and 6.25μM of peptide in 50mM Tris, pH 7.6 were constituted. The solutions were incubated for 20 min at 4º Cbefore measuring their activity using phosphoglycerate kinase assay -1. The protein activity was measured spectrophotometrically by following the oxidation of NADH at 340 nm. The assay was performed at 20º C in a 3.04mL reaction mixture containing 100 mM triethanolamine hydrochloride buffer (adjusted to pH 7.6 with KOH), 40 mM ATP, 150 mM 3-P-glycerate, 12 mM NADH, 100 mM magnesium acetate, 40 mg/mL bovine gamma globulin and glyceraldehyde 3-phosphate dehydrogenase (crystalline suspension in 3.2 M (NH4)2 SO4 solution that is about 10 mg/mL enzyme at 80 U/mg). The reaction was initiated by the addition of 20 uL of enzyme. PGKB:peptide complexes were subjected to G-75 Sephadex size-exclusion chromatography using FPLC, to study the binding. Absorbance of each fraction collected was measured spectrophotometrically at 280 nm and enzyme activity of fractions monitored using assay -1. Since 11-mer peptide does not have any aromatic residues absorbance was additionally monitored at 230nm in sample with this peptide.
Table 1: Leishmania mexicana C-terminal extension
| Peptide | Sequence |
|---|---|
| 11-mer | 418VKGRGPLLKCA428 |
| 18-mer | 430GGASPSNESCPRRREGIW447 |
| 25-mer | 451FIVTEIVKLVGALLIGIFIGRRLNT479 |
The entire sequence of the 63-mer C-terminal extension is represented by:
417EVKGRGPLLKCACGGASPSNESCPRRREGIWGGGFIVTEIVKLVGALLIGIFIGRRLNTKLIR479
Effect of peptides on substrate binding to PGKB
The 3 peptides derived from C-terminal extension of PGKC were incubated with PGKB at the ratio of PGKB: peptide, 1:1, for 20min at 4º C and tested for the effect on affinity for ATP and 3-PG using phosphoglycerate kinase assay- 1. The PGKB: peptide (6.25 μM each) mixtures were prepared in 50mM Tris and 0.15M NaCl buffer, pH 7.6, and incubated for 20 min at 4º C. ATP and 3-PG binding was studied at varying concentrations of the substrates and Michaelis Mentin constants, Km and Vmax obtained. All data represented were replicated at least thrice.
Leishmania culture supernatant assays
For culture supernatant experiments, L.donovani cultures were used which were a kind gift of Professor Madhubala, Jawaharlal University, New Delhi. PGK activity of the Leishmania donovani culture supernatants were monitored every 24 hours till 96 hours. PGK activity peaks at 72 hours in culture supernatants whereas total protein expression peaks at 48 hours. The 72 hour cultures were chosen for studies with peptides and the potent inhibitor of PGK, suramin. Stock solution of 0.1mM peptide and 40µM suramin were used in the inhibition studies.
Lipid interaction of protein:peptides complexes
Lipid vesicles were made by mixing chloroform dissolved phosphatidylcholine and phosphotidylethanol (10mg lipid in 1mL solvent) and drying over N2 gas overnight in desiccator. The lipid mixture was suspended in aqueous buffer, 50 mM Tris, 5 mM EDTA, pH 7.5, and sonicated till a clear or almost clear suspension was obtained. First PGKB_Lmexicana was added to the vesicles and incubated 4 hours at 4º C and then stock solution of peptide was mixed with vesicles and resonicated to get a clear solution. Final composition was 1:1:5 Protein:peptide:lipid. Gel-filtration chromatography with peptides was performed on a Sephadex G-25 (medium) column (1.5 x 30 cm) either before or after incubation with lipid vesicles and the fractions were similarly monitored for peptide and lipid by UV absorbance and scattering at 600nm respectively.
Homology modelling using SWISS-MODEL Server
The primary sequence of PGKC from the Leishmania mexicana contained in Swiss-Model template library (SMTL) was used as input in template search with BLAST [19] (SMTL, last update: 2014-12-12, last included PDB release: 2014-12-05). A total of 42 templates were found. Template search with HHBlits was done using the procedure outlined in [20] and 43 templates found. The template with the highest quality was selected for model building. Models are built based on the target –template alignment using ProMod-II. Coordinates which are conserved between the target and the template are copied from the template to the model. Insertion and deletions are remodeled using a fragment library. Side chains are then rebuilt. Finally, the geometry of the resulting model is regularized by using a force field. In case loop modelling with ProMod-II [21] does not give satisfactory results, an alternate model is built with MODELLER [22]. Homology model was built for the segment containing residues 6-415 of PGKC_Lmexicana using the most homologous structure as template, Phosphoglycerate kinase C from T. brucei (PDBID 16PK, [23]. The homology modeling of the 63-residue Cterminal extension was done using the structure of BCL2/Adenovirus E1B 19kDa protein-interacting protein 3 (PDBID 2KA1, [24]. The global and perresidue model quality has been assessed using the QMEAN scoring function [25]. A QscoreOligomer is calculated and the oligomeric state of the target is predicted to be the same as in the template when QscoreOligomer is higher or equal to 0.5 [26].
Secondary Structure Prediction using JPred
JPred [27] software was used to independently predict individual fragments secondary structures of individual fragments of PGKC_Lmexicana using the FASTA sequence as input. However, the data from the SWISSMODEL Server alone was used in the case of PGKC (6-415). For the 63-mer C-terminal extension (i.e., residues 417-479) JPred data was used as secondary structure prediction. This data was used in Chimera 1.10.1 software (see later) to make a tentative model for the whole PGKC_Lmexicana (residues 1-479).
Modelling of the interactions between the PGKC (6-415) and C-terminus (417 –479) using ClusPro 2.0 Protein-protein docking software and Chimera
Protein-protein docking was done using ClusPro server [28]. The structural relationship between the core that is 6-415 of PGKC_Lmexicana (receptor) and 63-mer C-terminus extension 417-479 of PGKC_Lmexicana (ligand) is analysed from graphs which show the clustering of docking models depicting RMS versus predicted energy of interaction. ClusPro uses 3 steps to finalize the output docking conformations, rigid body docking, fast Fourier transform (FFT) and energy minimization to establish structures that are stable over time. Structures with low Gibbs free energy fall into clusters and within this cluster those with low RMSD values are selected. This lead to 40 models that fall at the centre of each cluster from four energy criteria, Van der Waals, hydrophobic, electrostatic and balanced coefficients. The models for complexes built by ClusPro 2.0 were visualized using PyMol software [29]. All interactions between receptor and ligand pertaining to substrate active sites were analysed in each of the models obtained from the four energy criteria listed above. The interacting distance between two residues was set to a maximum of 5.5Å.
Chimera 1.10.1 and Chiron/GAIA Server
Chimera was used to generate the final structure of whole PGKC_Lmexicana (1-479). CHIRON molecular dynamics software was used to minimize the structure to find favourable energetics and to remove severe clashes. ((http://chiron.dokhlab.org). The protein structure was validated using GAIA [30,31].
Results and Discussion
Peptide interaction with PGKB
Three possible functions of the 63-residue C-terminal extension of PGKC_Lmexicana has been suggested, a) association with other proteins (soluble or glycosomal membrane bound), b) ligand binding and c) modulation of enzyme activity [32]. Although studies with whole proteins and deletion mutants can give insight into the role of this extension in enzyme activity, we were more interested to find out how the 63-mer extension interacts with rest of the sequence of PGKC_Lmexicana. Towards this goal, the interaction of 63-mer extension derived peptides were tested with PGKB_Lmexicana as Leishmania PGKB and PGKC share a high sequence similarity. Semi-synthetic protein complexes containing one natural and one synthetic fragment have been classically used in the study of interacting residues in protein folding [33]. Since PGKB_Lmexicana itself does not have the 63-mer extension, we used it to mimic the interactions of the C-terminal extension within PGKC_Lmexicana. The sequences of the peptides derived from the 63-mer are shown in Table 1 as 11-mer, 18-mer and 25-mer. A completely unrelated peptide (kind gift of Dr. R. P. Roy, NII) was used as control in all experiments. PGK is known to spend most of the folded conformation in the thermodynamically favourable open form. It was not surprising to find that the 63-mer extension peptides when incubated with purified PGKB_Lmexicana showed an interaction. The interactions were strong and unambiguous. The peptides were used to study a) Lipid interaction b) Interaction with PGKB_Lmexicana and c) In vivo effect of peptides on Leishmania cultures. The data corresponding to PGKB_Lmexicana interaction are now described. Figure 1A shows a typical enzymatic assay profile with PGKB_Lmexicana represented by the solid circles. Two of the 3 peptides, 11mer and 25-mer showed a strong interaction (µM range) with PGKB_Lmexicana (Figure 1A, B and C). Figure 1A shows the real time assay at 1:1 PGKB:peptide concentration. Enzyme activity is slightly enhanced by 11-mer (bottom most curve); 18-mer did not affected PGKB activity; 25-mer strongly inhibited enzyme activity (topmost curve). Figure 1B shows the same data in terms of relative activity where free PGKB_Lmexicana is represented as 100%. The enzyme activity of the PGKB:18-mer complex remains close to activity of free PGKB_Lmexicana. PGKB:11-mer complex is slightly more active than free PGKB_Lmexicana at higher concentrations of peptide.
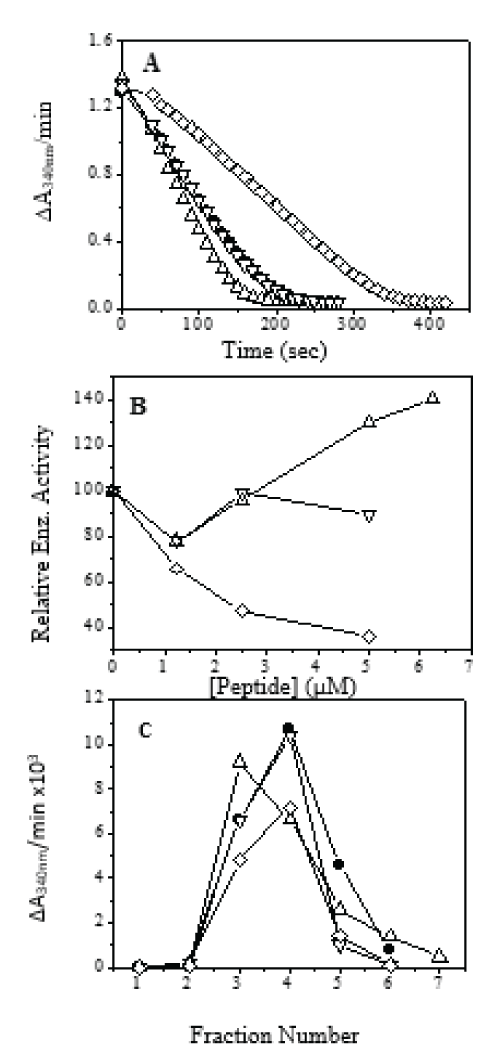
Figure 1: Effect of peptides on PGKB_Lmexicana activity: -â—- PGKB, -∆- PGKB+11mer, -- PGKB+18mer, -ï‚£-PGKB+25mer. (A) Effect of 1:1 ratio of peptide to protein on PGKB activity. (B) Relative enzyme activity of PGKB with different peptides. (C) Gel filtration chromatography on Sephadex G-25 of protein/peptide complexes: -â—- -PGKB, -∆- PGKB+11mer, -- PGKB+18mer, -ï‚£-PGKB+25mer.
PGKB:25-mer shows lower activity than PGKB_Lmexicana. All the PGKB:peptide complexes were passed through a gel-filtration chromatographic column and the enzyme activity measured for each of the fractions (see Materials and Methods). The PGKB:11-mer shows reduction in enzyme activity (Figure 1C) and shift in the elution volume compared to free PGKB_Lmexicana elution profile (shown in solid circles in Figure 1C).
PGKB:18-mer elutes at the same position as free PGKB_Lmexicana and the activity profile exactly overlaps that of the free enzyme. PGKB:25-mer shows a reduced peak height indicating inhibition of activity with strong binding. The shift in elution Vmax in the PGKB:11-mer complex, is indicative of a change in the stokes radii or the conformation of the protein. We conclude that both 25-mer and 11-mer bind strongly to PGKB_Lmexicana. Using standard PGK assay (see Materials and Methods), we obtained the Michaelis Mentin constants for ATP and 3-PG in the presence of the peptides, (Figure 2 and Table 2). 11-mer peptide showed a decrease in the Km value for both ATP and 3-PG binding to PGKB but the difference is significantly more with the triose substrate 3-PG, indicating that the binding affinity of PGKB_Lmexicana for 3-PG is enhanced seven times approximately by the 11-mer peptide. This could be the reason for enhanced activity of PGKB_Lmexicana in the presence of 11-mer (Figure 1A, 1B). Enhancement of PGK activity has been seen previously with low concentration of small anions and has been attributed to an allosteric effect. Such a large change on 3-PG binding by 11-mer peptide in the PGKB:11-mer complex may be affected through a conformational change in the protein. Recently it has been reported that a hydrophobic patch is buried in the core of the PGK protein in the open conformation, which gets exposed on formation of the closed catalytically active conformation [34]. Pioneering research in the biochemistry of PGK has come from the laboratory of Maria Vas: they have shown that the highest conformational change in PGK happens on the binding of triose substrates, 1,3-bisPG followed by 3-PG [35].
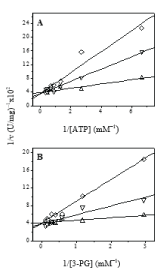
Figure 2: Steady-state kinetic plots: -∆- PGKB+11mer, -- PGKB+18mer and -- PGKB+25mer. Kinetic assays were carried at varying concentration of either ATP (A) or 3-PG (B).
Table 2: Binding of ATP and 3-PG to PGKB_Lmexicana in absence and presence of peptides
| Enzyme Complex ↓ Substrate → | ATP (Km values in mM) | 3-PG (Km values in mM) |
|---|---|---|
| PGKB | 0.24 | 1.45 |
| PGKB + 11-mer | 0.16 | 0.21 |
| PGKB + 18-mer | 0.33 | 1.18 |
| PGKB + 25-mer | 1.74 | 1.57 |
This conformational change is necessary for the next step of catalysis. The increased affinity for 3-PG may be due to 11-mer induced conformational change in PGKB_Lmexicana. In contrast to the 11-mer peptide, the 25mer peptide showed an increase in the Km value for both ATP and 3-PG binding to PGKB_Lmexicana but in this case the increase was much greater for ATP.
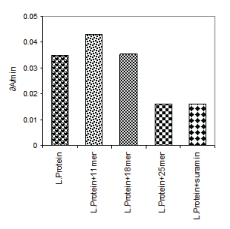
The several fold decreases in affinity for ATP substrate explains the inhibitory effect of 25-mer peptide on enzyme activity described earlier (Figure 1). The 25-mer peptide seems to sterically interfere at the ATP binding site. These data are used basis to generate a model of PGKC_Lmexicana as discussed later. The binding of the 11-mer, 18-mer and 25-mer peptides to PGKB_Lmexicana could be reproduced in in vivo in studies with Leishmania donovani cultures (Figure 3). Interestingly suramin, the known inhibitor of PGK, has the same inhibitory effect on PGKB_Leishmania as 25-mer peptide.
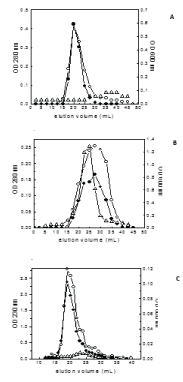
Finally the interaction potential of the three peptides with lipid vesicles in the presence of protein (PGKB_Lmexicana) were monitored by passing PGKB:peptide:PC/PE complexes through a gel filtration chromatography (Figure 4). 25-mer and 18-mer have aromatic residues and can be monitored by absorbance at 280 nm. Since PGKB will contribute to absorbance at 280 nm, it was monitored by enzyme activity. 11-mer was monitored by peptide backbone absorption at 230 nm. PGKB:25-mer:PC/PE (Figure 4A) elution profile shows lipid co-eluting with 25-mer peptide at elution volume 19-20 mL. PGKB protein elution volume is seen to be a broad peak and shifted far with a slight maximum at 28 mL. This shows that in the presence of both lipid and PGKB_Lmexicana, 25-mer has a clear preference for lipid over PGKB_Lmexicana. In the case of PGKB:18-mer:PC/PE (Figure 4B) the elution maximum for PGKB_Lmexicana (23-24 mL) is slightly shifted from the elution maximum for peptide (28 mL). Lipid scattering is seen as a broad peak with two maximum. Thus 18-mer has a weak if any interaction with PGKB_Lmexicana and no interaction with lipid. Similar to 25-mer, 11-mer shows co-elution with lipid at elution volume 18 -19 mL. The protein activity has a maximum at 23-24 mL showing some interaction with peptide and indicating a slow exchange between PGKB:11-mer and 11-mer:PC/PE. We therefore suggest that the 25-mer region of 63-mer C-terminal extension binds to the lipid (in the cell it will be the glycosomal membrane) to form a transmembrane helix or binds to the PGKC_Lmexicana near the nucleotide binding site in a mutually exclusive manner. 11-mer binds to protein and lipid simultaneously. It is possible that 11-mer being hydrophobic in nature is binding at a hydrophobic patch on PGKB_Lmexicana. One possibility is the hydrophobic patch mentioned by Zerrad et al [34] which becomes exposed in the closed conformation of PGK but remaining buried in the open form. In the case of T.brucei the truncated PGKC without C-terminal extension showed reduction in affinity of enzyme for 3-PG without affecting its affinity for ATP [36] suggesting that the extension enhances 3-PG binding. Overall the results indicate that the 63-mer extension in PGKC affects the activity as well as the binding of substrates, and may also bind to the glycosomal membrane.
SWISS Homology Modelling
Leishmania mexicana PGKC was found to be most homologous to the Trypanosoma brucei phosphoglycerate kinase (PDBID 16PK). The sequence identity was 75.5%. Figure 5 shows the 3-dimensional ribbon diagram of residues 6-415 of PGKC_Lmexicana obtained by homology modelling. The last 63 residues are not included as they are unique to Leishmania PGKC. The global and per-residue model quality has been assessed using the QMEAN scoring function [25]. GMQE of 0.76 and QMEAN4 value of -1.17 were obtained, indicating monomeric structure. The α/β motif in both domains and the Rossmann fold in the C-domain were noted and the main hinge region highlighted in the structure. No secondary structural element is involved in interconnecting the domains. The second hinge involving the terminal residues fold into helix 15 in the N-domain (Figure 5). Thus all structural features are as expected for regions 6 – 415 in PGKC_Lmexicana.
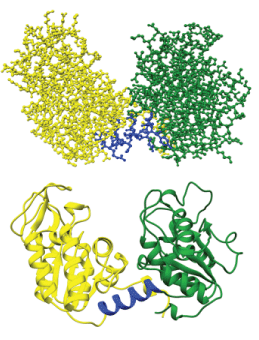
Figure 5: Homology modelling with SWISS-MODEL. The figure shows two versions of the same enzyme; a ribbon view with ribbons depicting alpha helices and arrows indicating beta strands and a ball and stick view depicting atoms of the peptide backbone and their respective side chains. Represented in green is the N-terminal from residues 6-187, in blue is the hinge region from residues 188-201 and in yellow is the C-terminal from residues 202-415.
The result of the homology modelling of the 63 residue C-terminal sequence (sequence as in Table 3) submitted to SWISS-MODEL (63r_PGKC_Lmexicana) is shown in Figures 6a and 6b. The 63 residue target sequence was searched with BLAST (19) against the primary amino acid sequences contained in SWISS-MODEL template library for evolutionarily related structures matching the target. A total of 10 templates were found with at least 25% overall sequence identity and maximum 40% sequence identity to residues 35-59 of 63-mer C-terminal extension of PGKC_Lmexicana. The QMEAN4 value is -1.07 and GMQE of 0.25. Overall 13 templates were used to generate the model. 6 of the 13 templates used the BCL2 adenovirus E1B PIP3 protein and indicate a homodimer whereas the remaining 7 templates indicate a monomer. EIB PIP3 exists as a homo-dimer and contain a GXXXG motif what has been found to be involved in dimerization [24]. Figure 6a shows a homodimer of residues 35FIVTEIVKLVALLIGIFIGRRLNT59 in the 63-mer region. The single long helix is consistent with our earlier studies of NMR structural determination of the peptide (35FIVTEIVKLVGALLIGIFIGRRLNT59) in SDS micelles [16].
Table 3: Amino acid sequence of Leishmania mexicana PGKC. Important residues are italized in bold.
| Domain | Residues |
|---|---|
| N-terminal | 1MSLVLKKSIDDVALKDKKVLIRVDFNVPVKNGEITNDFRIRSALPTIQKVLKEGGSCILM SHLGRPKGARMSDPKPEKGVRGYEEAATLRPVAAALSELLEKKVAFAPDCLNASIYVSKL KRGDVLLLENVRFYTEEGSKKEEEADAMAKVLASYADLYVSDAFGTAHRDSATMTGIPKV LGSGYAG187 |
| Hinge region | 188YLMEKEINYFSRVLN203 |
| C-terminal | 204NPPRPLVAIVGGAKVSDKIELLDNMLGRINYLVIGGAM AYTFQKAQGRKIGISMCEEDKLDLAKSLLKKAQERGVQVLLPVDHVCNKEFKAVDSPLVT EDVDVPDGYMALDIGPKTIHMYEEVIGRCKSAIWNGPMGVFEMPCYSKGTFAVAKAMGTG TQKDGLLSIIGGGDTASAAELSGEAKNMSHVSTGGGASLELLEGKTLPGVAILTDK416 |
| 63-mer C-terminal extension | 417EVKG RGPLLKCACGGASPSNESCPRRREGIWGGGFIVTEIVKLVGALLIGIFIGRRLNTKLIR479 |
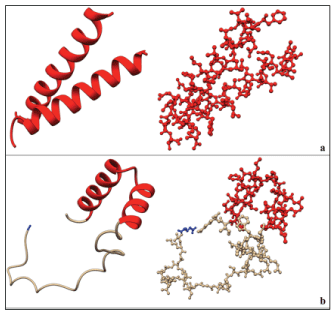
Figure 6: a) Homology modelling with SWISS-MODEL. Two forms of the same structure are shown, on the left is a ribbon representation while on the right is a ball and stick representation. The figure shows a homodimer of residues 35FIVTEIVKLVGALLIGIFIGRRLNT59. This is shown in red. This structure is present in the BCL2 adenovirus E1B PIP3 protein with a 40% sequence similarity. b) Modelling of the 63-mer in Chimera 1.10.1 using JPred secondary structure prediction data. In this case the alpha helix is a monomer having the residues 35FIVTEIVKLVGALLIGIFIGRRLNTKL61 (see text for details). In blue is the first residue of the 63-mer which is a glutamic acid.
In order to get the structure of the entire PGKC_Lmexicana we needed a model for the 63-mer extension (Table 3). We built the 63-mer sequence using JPred data and the result of subsequent homology modelling is shown in Figure 6b. In red is the same region of helix (35FIVTEIVKLVALLIGIFIGRRLNT59) but in this model the helix is interrupted with a bend. The bend in the helix is at residues 46-49 which was more solvent exposed than the rest of the helix. Glycine at position 50 is predicted to have 0% solvent accessibility, thus being completely buried. In the in vitro environment of SDS micelles the 25-mer solubilizes completely and gives one long helix without bent [16]. These data seem to indicate that the region of the helix in the 63-mer extension can undergo conformational transition based on the environment. The rest of the 63-mer seems largely unstructured although our circular dichroism data showed a helix in the region of 11-mer that is 418VKGRGPLLKCA428.
ClusPro Protein-Protein Docking Server
The orientation of 63-mer C-terminal extension in the geoscape of PGKC_Lmexicana was obtained using Cluspro 2.0 Protein-Protein Docking and visualized in PyMol Molecular Graphics Software (see Materials and Methods). In the protein-protein docking predictor PGKC_Lmexicana (6-416) or core protein is treated as the receptor and 63-mer domain as the ligand (sequencing and numbering shown in Table 3). The software filters possible orientations in terms of surface complementarities of any two protein complexes. The initial docking gave us about 125 models for analysis. These were visually analysed taking into account biochemical data on interaction between PGKB_Lmexicana and peptides from 63-mer.
First the position of the 63mer in relation to the core protein that is 6-416 of PGKC_Lmexicana henceforth called receptor, was analysed and viewed using PyMol. Secondly the interacting residues of the receptor and ligand that have a distance of less than 5-5.5A between them in space were identified. Third the distances between residues of the ligand near to the 3-PG, ATP active sites and hinge regions of the receptor were found. Models were analysed based on these criteria.
There were three possible orientations of the receptor and ligand in space. A vertical conformation in which the ligand passes through the receptor vertically splitting the receptor in two domain. Second, a horizontal conformation in which the ligand passes through the receptor on the X-axis plane. In both of these the TMD region interacts with the receptor. The third is a sigmoidal conformation of the ligand running the face of the receptor with the TMD tethered away from the receptor: this was rejected because it was not supported by our biochemical data.
Strikingly, majority of the docked conformations show that the ligand interfaced with the receptor exactly in the transmembrane helix region and slightly prior, that is residues R443 – N474 (for numbering see Table 3) at distances less than 5.5Å to backbone atoms. The results are shown as histograms in Figure 7. We see that R39 from the triose binding pocket of receptor comes close with ligand (63-mer extension) and the 63-mer extension residues interacting are most often L49 followed by G51 (number as shown in figure legend of Figure 7). In the balanced coefficient model we find ligand approach to the basic patch R169 and H168 on receptor at distances of 6Å and 9Å respectively. In another instance in the electrostatic model we found ligand approach to H62 of receptor at close to 9Å. In a comparison of known crystal structures of PGK Vas et al., [7] find that H168 is involved in electrostatic inter-domain interaction.
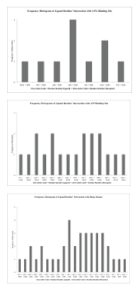
Figure 7: 1EVKGRGPLLKCACGGASPSNESCPRRREGIWGGGFIVTEIVKLVGALLIGIFIGRRLNTKLIR63 Frequency histogram using ClusPro 2.0 Protein-Protein Docking Server of residue interactions between the 63-mer ligand with, a) the 3-PG binding site comprised of the residues R22, R39, H62, H168 and R169 of the receptor; b) ATP binding site comprised of the residues L312, N335, G339, V340, E342 and D374 and c) hinge region comprised of the residues Y188 -L201 of the core protein, analysed in Chimera 1.10.1.
Approach of 63-mer extension to the ATP active site region of receptor is frequent in all the models analysed: the interacting 63-mer extension residues fall in the region of the 25-mer peptide of our biochemical studies. In the nucleotide binding pocket of phosphoglycerate kinase, the adenine ring is completely buried in a pocket lined with mostly hydrophobic residues. The ribose part binding on a shallow depression and interacts with glutamate E342. Out of all the ATP binding residues within the PGKC_Lmexicana core protein only two are outward facing, E342 and D374; 63-mer extension approaches the vicinity of these two residues. The 63-mer extension residues involved are L43, V45, I54, G55 and R56. In all the models there is no proximity of ligand to the adenine ring binding hydrophobic pocket and the minimum distances seen were 15Å.
In many models we see the hinge region residues Y188, L189, and K192 of PGKC_Lmexicana approach the 63-mer extension, especially in hydrophobic or balanced coefficient models. These residues form part of strand βL. The ligand also come in its vicinity to T393 of receptor which, interacts with the carboxylate of 3-PG and is in proximity to triose site triple glycine, G394 – G396 [7]. A large number contacts to inter-domain residues S377, T393 are seen with 63-mer extension. The residues S377 is close to triple glycine at G371-G373, the interaction being important in the closed conformation of PGK. These triple glycine have an important role to play in interaction to the transferring phosphate [37]. The region 50GIFIG54 of 63-mer extension is prominent in all the above mentioned interactions.
The reader may be reminded that the 25-mer peptide within the 63-mer extension can bind either to lipid membrane or PGKB_Lmexicana in our biochemical studies but not both at the same time (Figure 4). The 50GXXXG54 motif within the 25-mer region (466GIFIG470 in the whole protein sequence) is a potential transmembrane dimerizing motif. However no experimental data to date exists on whether PGKC forms a dimer in the glycosomal membrane. The observation through docking suggest that the ligand (63-mer extension of PGKC_Lmexicana) approaches the nucleotide binding residues E342 and D374 of receptor (PGKC_Lmexicana) via the TMD region (residues 451-470 of PGKC_Lmexicana, Table 3). The 63-mer extension folds in a region of PGKC_Lmexicana where it can interact with the hinge as well as position between the N-domain and C-domains (histogram, Figure 7). The template of T. brucei PGKC which has been used to model PGKC_Lmexicana is in a closed conformation. It appears that 63- mer C-terminal extension of PGKC_Lmexicana finds a cleft between the two domains and that 50GIFIG54 motif in the 63mer domain makes numerous contacts with the core of PGKC_Lmexicana. A tentative model based on over 125 models generated by ClusPro with all of the properties discussed so far, is presented below.
Generating a model for PGKC_Lmexicana (1-479)
The model of PGKC_Lmexicana considered all the practical and theoretical results discussed so far. Initially Chimera 1.10.1 was used to join the homology model PGKC_Lmexicana (6-416) and the 63-mer C-terminal extension through a peptide bond. A model for PGKC_Lmexicana (6-479) was generated, shown in Figure 8a. This model complies with all of the biochemical data, the proximity between residues found from ClusPro 2.0 as well as general rules for protein folding. Model, M0, is tailor made to fit the biochemical data, in particular the proximity of 25-mer region (sequence shown in Table 3) to E342. All steric clashes in the structure were resolved through energy minimization. The model M0 generated through chimera is compared to the chimera minimized structure M1, which was further minimised using Chiron resulting in the final structure M2 and is compared in Figures 8a. The RMSD value for each residue pair in M0 as compared to M1 is 0.284 with a high Q-score of 0.991. However the structure minimized and validated using Chiron and GAIA, (both online servers) respectively had a RMSD of 0.299 and a lower Q-score of 0.726. According to the GAIA report, model M1 had issues with steric clashes, protein geometry, side chain integrity and unresolved hydrogen shells in the core and side chain of the protein. The software itself resolves these issues and generates a model that falls within the normal distribution for a set crystallized protein structures. The steric clashes in M1 resulting in high clash energy fell at the end of a distribution curve of clash energy/contact of crystallized protein structures. After automatic correction of structure through minimization, this clash energy falls within the normal distribution (Figure 8b). Minimization helped to bring the protein geometry into the Gaussian distribution of known protein structures on these criteria. The clash energy in terms of VdW repulsion energy dropped from 299.992 kcal/mol to 133.311 kcal/mol in the final model which equates to a clash ratio of 0.0178. This value falls within the normal distribution and has a p-value of 0.581. The final resolved and refined structure is model M2 that we wish to present as a tentative structure of PGKC_Lmexicana. The solvent accessible surface area in M2 was within normal distribution with a p-value of 0.804. We also confirmed the angular data from the Ramachandran Plot.
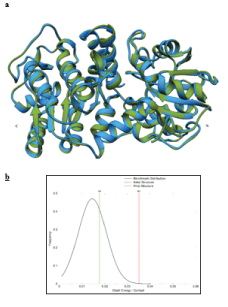
Figure 8: a) Represents the comparison between Model M1 (green) and M2 (blue) with the N and C terminals labelled. b) The chart shows the results of the minimization of model M1, resulting in model M2. The clash energy expressed as the VdW repulsion energy falls within the normal distribution represented by the yellow line, labelled as M2.
Folded conformation of PGKC_Lmexicana
A critique and evaluation of the validity of the final model M2 (Figure 9) is presented here. The key questions to take into consideration are: 1) is the structure viable 2) do we see the interactions of the 63-mer C-terminal extension with the core of PGKC_Lmexicana in accordance with the peptide studies and suggested by ClusPro 2.0 data.
Regarding the validity of the structure we use the accepted rules for protein structure and folding. As mentioned above, minimization brought the structure within the normal distribution of structural parameters in the database of globular proteins with known crystal structures. As PGKC_Lmexicana is not globular but a two-domain protein some discrepancy is noted in the number of unsatisfied hydrogen bonds in the core compared to the dataset. However this discrepancy is not there for the hydrogen bonds in the shell of the protein that interact with the environment. Further our model M2 shows a normal distribution of solvent accessible surface area. As far as protein geometry is concerned the following discrepancies were there; a) bond length for residue 462:CA---462:CB is 1.75Å whereas the maximum accepted length is 1.69Å b) 32:N---32:CA---32:CB bond angle is 128º whereas maximum accepted is 120º c) 409:C---409:CA---409:CB angle is 127º whereas maximum accepted is 118º and 411:N---411:CA---411:CB is 139º where maximum allowed is 120º. The areas of discrepancy seen are either at the chain termini or in the unstructured part of 63-mer extension and therefore not considered significant.
Next comes the point regarding our biochemical studies whose main points are that a) 25-mer region of the 63-mer C-terminal extension inhibits ATP binding of PGKB_Lmexicana through direct interaction b) 11-mer region of the 63 residue C-terminal extension allosterically enhances 3-PG binding and c) both 11-mer and 25-mer interact with lipid and may assist for membrane localization of PGKC_Lmexicana. d) Lastly, the closeness of the 25-mer region of 63 residue C-terminal extension to the ATP binding site residues seen in our ClusPro 2.0 docking studies.
Figure 9 shows part of the 63-mer extension shown as two helices is interacting within the groove between the two domains of the protein; this part corresponds to the 25 mer peptide (of our earlier NMR studies) that is 450FIVTEIVKLVGALLIGIFIGRRLNTK471. The beginning of the extension which is 11-mer and 18-mer is mostly unstructured is positioned at the base close to the N-domain. The observed conformation of PGKC_Lmexicana is closed, same as the T. brucei PGKC. The ClusPro 2.0 results showed 25-mer peptide close to E342 and D374, both part of the ATP binding site at a distance of <5.5Å. Figure 10 shows 25-mer region approach E342: between Ile 455 – E342 at 5.39Å, Val 456 – E342 at 6.72Å and Gly 469 –E342 at 7.70Å. The distances refer to the carbon alpha atoms whereas the side chains may approach closer to E342 and thus exert their inhibitory effect of ATP binding. Interestingly G469 is the last glycine of 466GIFIG470 (that is GXXXG motif) helix to be close to E342 but not D374. The interaction of 25-mer region to triose site are shown in Figure 10,11. Arg 471 – Arg39 at 7.70Å, Arg471 – Phe38 at 8.51Å and Thr474 –Arg39 at 9.54Å (carbon alpha to carbon alpha) respectively. Our biochemical experiments show that the 25mer doesn’t affect 3-PG binding to PGKB_Lmexicana but the starting conformation in those studies was an open conformation unlike the partially closed structure of our model, PGKC_Lmexicana. The 11-mer is the region of the 63 residue C-terminal extension which is flexible and folds at the base of the N-domain. The proximity of 11-mer region to the N-domain as opposed to C-domain is consistent with interaction near the triose binding site.
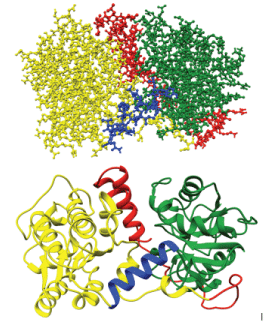
Figure 9: Model M2, the final tentative of PGKC_Lmexicana (residues 1-479). Represented in green is the N-terminal from residues 6-187, in blue is the hinge region from residues 188-201, in yellow is the C-terminal from residues 202-415 and in red is the 63-mer C-terminal extension (residues 416 – 479).
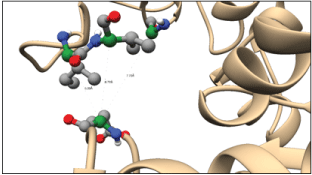
Figure 10: Interaction of the 25-mer fragment of the 63-mer C-terminal extensions with the ATP binding site residue E342. The residues shown in ball and stick representation form part of the helix that sits between the two domains of the enzyme. The interactions seen above are between Ile 455 – E342 5.39Å, Val 456 – E342 6.72Å and Gly 469 –E342 7.70Å. Green: alpha carbon, grey: carbon, blue: nitrogen, red: oxygen and white: hydrogen.

Lastly, Figure 12 shows that the region 462LLIGIFIG469 is part of a large hydrophobic patch on the surface of PGKC_Lmexicana. L462, L463 and I464 residues form an exposed hydrophobic region on the surface. This region could be attracted to the glycosomal membrane to allow for membrane insertion and transport. The largest known hydrophobic patch in any PGK is about 600Å2 in the nucleotide binding cleft. The second largest of about 400 Å2 lies in the N-domain beside the phosphoglycerate binding loop [38]. The new hydrophobic patch seen by us (Figure 12, bottom) is more globular than the linear shape seen in the other two patches. Although abundant, GXXXG motifs are not the only transmembrane homodimerization signals and some may be functionally insignificant [39]. In the case of the human proton-coupled folate transporter, the GXXXG motif is important for protein stability in the transmembrane domain and mediates intramolecular packing [40] rather than protein/protein interaction. Therefore this domain in PGKC_Lmexicana could have a similar role
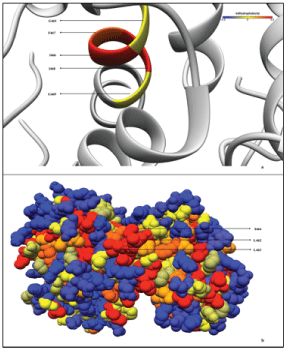
Figure 12: Hydrophobicity map of the PGKC_Lmexicana model structure. The Kyte-Doolittle hydrophobicity [44] of the membrane interacting GXXXG motif part of the 25-mer C-terminal extension is shown on the top panel. Bottom panel shows the hydrophobic residues that probably help the GXXXG motif localize into the membrane.
Conclusion
PGK is one of the oldest enzymes in evolution. It was in 1979 that the X-ray crystallographic structure of horse muscle PGK was determine and a hypothesis of a possible hinge bending enzyme revealed [37]. Since then many PGK structures have shown that in spite of amino acid differences the tertiary structure is conserved. The ï¡/ï¢ tertiary structure, the active site residues and many inter-domain interactions are notably preserved characteristics. The tertiary structure being conserved, the thermodynamic stability of various PGK’s however varies. In our own laboratory, preliminary queries of protein unfolding behaviour has shown the Leishmania and Yeast PGK’s differ significantly in their stability, the stability of the yeast enzyme being significantly higher (data not shown). Between the open and closed conformations of PGK, the former is more stable as it buries a hydrophobic patch that gets exposed upon transition to the closed conformation. Burial of these hydrophobic residues is thought to drive the protein back from catalytic conformation to the non-catalytic open conformation [34]. The dynamics of PGK is well established. Bowler [41], shows that the degree of openness of PGK may be even higher than observed in crystal structures with a 53 degree angle between the domains, and that Brownian forces influence the energetic landscape of conformations and the catalytic cycle. Previously the domain closure was thought to be mediated through electrostatic interactions between domain but it now appears that a spring like mechanism driven by a large hydrophobic patch works against Brownian and electrostatic forces to cause catalysis. The Brownian motion across the energy landscape that assist protein folding both in vitro and in vivo [42,43] can be controlled or altered through hydrophobic forces.
Our model of PGKC_Lmexicana is shown in Figure 9: the end part of the 63-mer C-terminal extension positioned in the cleft between the N- and Cdomains of PGKC, which includes the 25-mer section (435FIVTEIVKLVGALLIGIFIGRRLNTK476). This 25-mer section has the property of either binding to residues in the nucleotide binding site or forming a single helix in SDS micelles called the transmembrane segment (TMS, [16]), these two interactions being mutually exclusive. The first half of 63-mer Cterminal extension that is residues 417E--to--P439 is mostly flexible although short stretches of helix and strand are present, and it lies in the general region of the N-domain. Within this region is the 11-mer peptide 417VKGRGPLLKCA442 that allosterically influences 3-PG binding and also has a weak potential to form membrane bound helix, these two interactions are able to take place simultaneously. The conformation of PGKC_Lmexicana represents a closed conformation. An important feature of the model is an exposed hydrophobic patch formed by 462LLIGIFIG469 of 63-mer extension; this motif, gets buried as transmembrane segment in the structure obtained in SDS micelles by NMR [16]. This may be important trigger point for interaction with the glycosomal membrane. The helix which corresponds to TMS is a discontinuous helix in the model shown in Figure 9. Discontinuous helices are often found in transporter proteins [44]. Furthermore, GXXXG motif such as is present in our case may have function in stabilizing the protein conformation given the sequence context such as the presence of neighbouring β-branched residues [18]. In our case the neighbouring β- branched residues are at positions V460 and I465. Thus, the structural importance of the hydrophobic patch and GXXXG may be in stabilizing the protein conformation when there is no membrane interaction. Since Quinones et al., [45] found a 50 kDa protein (phosphoglycerate kinase C) the most abundant protein in the glycosomal membrane of L. Mexicana, obviously, in vivo there is an interaction of PGKC with the glycosomal membrane. We too have made the observation that when PGKB_Lmexicana and PGKC_Lmexicana are incubated with lipid vesicles (1:1 protein:PC/PE) and passed through a gelfiltration chromatography column (Sephadex G100), PGKC_Lmexicana coelutes with lipid suggesting binding to the lipid whereas PGKB_Lmexicana does not co-elute (Data not shown). The question that begs answer is whether this interaction of PGKC_Lmexicana in the glycosome is transient or permanent.
Hydrophobic patches are loosely defined as regions of high hydrophobicity which may be lined by polar residues in some cases. The larger the size of the patch the more clear is its biological significance as in the case of the hydrophobic surface in the nucleotide binding cleft of phosphoglycerate kinase [38]. The smaller patches may have a more structural role in a protein. One such patch is found in inter-domain contacts of open conformation of porcine PGK (Fig 6D of reference 7). The hydrophobic patch that is formed by 462LLIGIFIG469 may represent the localized epistatic interactions controlling the evolvability of PGKC_Lmexicana. As shown in the case of the study of proteins of known tertiary structure from a phylogeny of 25 mammalian species [46] protein robustness during evolution is effected by the presence of pair-wise interactions between residues. PGKC_Lmexicana is an example of a protein that has evolved to acquire an entire new domain that is the C-terminal 63-mer extension while retaining its essential biological function. Already we have cited examples where PGK interaction with membrane and membrane proteins have been found. The dramatic change in the environment of PGKC_Lmexicana brought about by encapsulation in the glycosome will put structural pressure on the enzyme. In the well-studied case of HIV-1 protease, epistatic interaction in mutation covariance cause high evolvability and drug resistance of the protease [47]. Epistatic interaction help a protein stabilize during evolutionary pressures. The knowledge in this field is still scant and the exact role of epistatic pair-wise interactions or modularity on evolvability of function and robustness of PGKC_Lmexicana cannot be known at this time [48]. If encapsulation in the glycosome lead to a loss of thermodynamic stability of the protein then compensatory interactions would be required to re-stabilize it. Conceptually we can suggest a role of the hydrophobic patch and GXXXG in this.
Whether a switch type of mechanism for the 63-mer extension between insertion into the membrane with the rest of PGKC_Lmexicana in an open conformation to a closed conformation of PGKC_Lmexicana where the 63mer stabilizes this conformation is possible, remains to be investigated.
Furthermore, the factors leading to the evolution of structure and function of PGKC_Lmexicana, self-regulation, multi-functionality, and whether glycosomal PGK is itself the transporter of ATP and 3-phosphoglycerate across the membrane into the cytosol need to be uncovered. Studies in these directions have the potential to change how we think of metabolic regulation in Leishmania spp.
Acknowledgement
This study was supported by the National Institute of Immunology (NII), New Delhi and co-funded by the Centre for Scientific and Industrial Research, CSIR, New Delhi. We are grateful for the many suggestions by Dr. Debasisa Mohanty and Dr. R. P. Roy, NII, New Delhi, that have helped improve the manuscript. We are also deeply indebted to Professor Rentala Madhubala, JNU, New Delhi, for sharing the Leishmania donovani cultures.
References
- Ogino T, Iwama M, Kinouchi J, Shibagaki Y, Tsukamoto T, et al. (1999) Involvement of a cellular glycolytic enzyme, Phosphoglycerate kinase in Sendai virus transcription. J. Biol. Chem. 274:35999-36008.
- Boone TJ, Tyrrell GJ (2012) Identification of the actin and plasminogen binding regions of Group B Streptococcal Phosphoglycerate kinase. J. Biol. Chem. 287: 29035-29044.
- Cheng CP (2017) Host Factors Involved in the Intracellular Movement of Bamboo mosaic virus. Front Microbiol 8: 759. [crossref]
- Bakker BM, Michels PA, Opperdoes FR, Westerhoff HV (1999) What controls glycolysis in bloodstream form Trypanosoma brucei? J Biol Chem 274: 14551-14559. [crossref]
- Gualdron-López M, Vapola MH, Miinalainen IJ, Hiltunen JK, Michels PA, et al. (2012) Channel-forming activities in the glycosomal fraction from the bloodstream form of Trypanosoma brucei. PLoS One 7: e34530. [crossref]
- Achcar F, Barrett MP, Breitling R (2013) Explicit consideration of topological and parameter uncertainty gives new insights into a well-established model of glycolysis. FEBS J 280: 4640-4651. [crossref]
- Bernstein B.E, Michels PA, Hol WGJ (1997) Synergistic effects of substrate-induced conformational changes in phosphoglycerate kinase activation. Nature 385: 275-278.
- Vas M, Varga A, Graczer E (2010) Insight into the mechanism of domain movements and their role in enzyme function: Example of 3-Phosphoglycerate kinase. Current Prot and Pep Sci 11: 118-147.
- Cliff M (2010) Transition state analogue structures of human phosphoglycerate kinase establish the importance of charge balance in catalysis. J. Am. Chem. Soc. 132: 65076516.
- Jamdhade MD, Pawar H, Chavan S, Sathe G, Umasankar PK, et al. (2015) Comprehensive proteomic analysis of glycosomes from Leishmania donovani. OMICS, J. Integrative Biol. 19:157-170.
- Sommer JM, Peterson G, Keller GA, Parsons M, Wang CC, et al. (1993) The C-terminal tripeptide of glycosomal phosphoglycerate kinase is both necessary and sufficient for import into the glycosomes of Trypanosoma brucei. FEBS Lett. 316: 53-58.
- Subramani S (1993) Protein import into peroxisomes and biogenesis of the organelle. Annu Rev Cell Biol 9: 445-478. [crossref]
- Brocard C, Hartig A (2006) Peroxisome targeting signal 1: is it really a simple tripeptide. Biochimica, Biophysica Acta. 1763:1565-1573.
- Chowdhary G, Kataya AR, Lingner T, Reumann S (2012) Non-canonical peroxisome targeting signals: identification of novel PTS1 tripeptides and characterization of enhancer elements by computational permutation analysis. BMC Plant Biol 12: 142. [crossref]
- Reumann S, Buchwald D, Lingner T (2012) PredPlantPTS1: A Web Server for the Prediction of Plant Peroxisomal Proteins. Front Plant Sci 3: 194. [crossref]
- Kaushik S, Krishnarjuna B, Raghothama S, Aggarwal S, Raghunathan V, et al. (2012) Theoretical and in vitro studies of a C-terminal peptide from PGKC of Leishmania mexicana mexicana. Mol Biochem Parasitol 185: 27-35. [crossref]
- Lemmon MA, Treutlein HR, Adams PD, Brünger AT, Engelman DM (1994) A dimerization motif for transmembrane alpha-helices. Nat Struct Biol 1: 157-163. [crossref]
- Senes A, Gerstein M, Engelman DM (2000) Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with ß-branched residues at neighbouring positions. J. Mol. Biol. 296:921-936.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database research programs. Nucleic Acids Res 25:3389-3402.
- Remmert M, Biegert A, Hauser A, Söding J (2011) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9: 173-175. [crossref]
- Geux, N and Peitsch, MC (1997) SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modelling. Electrophoresis 18:2714-2723.
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779-815. [crossref]
- Bernstein BE, Williams DM, Bressi JC. Kuhn P, Gelb MH, Blackburn GM, et al. (1998) A bisubstrate analog induces unexpected conformational changes in phosphoglycerate kinase from Trypanosoma brucei. J.Mol.Biol. 279:1137-1148.
- Sulistijo ES, Mackenzie KR (2009) Structural basis for dimerization of the BNIP3 transmembrane domain. Biochemistry 48: 5106-5120. [crossref]
- Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343-350.
- Mariani V, Kiefer F, Schmidt T, Haas J, Schwede T (2011) Assessment of template based protein structure predictions in CASP9. Proteins 79 Suppl 10: 37-58. [crossref]
- Drozdetsky A, Cole C, Proctor J, Barton G.J. JPred4: a protein secondary structure prediction server. Nucleic Acids Research, April 16, 2015. Web Server issue.
- Kozakov D, Hall DR, Xia B, et al. (2017) The ClusPro web server for protein-protein docking. Nat Protoc 12: 255-278. [crossref]
- DeLano WL (2002) The PyMol Molecular Graphics System. http://www.pymol.org Version 1.2
- Ramachandran S, Kota P, Ding F, Dokholyan NV (2011) Automated minimization of steric clashes in protein structures. Proteins 79: 261-270. [crossref]
- Kota P, Ding F, Ramachandran S, Dokholyan NV (2011) Gaia: automated quality assessment of protein structure models. Bioinformatics 27:2209-2215.
- Adje CA, Opperdoes FR, Michels, PAM (1997) Organization, sequence and stage specific expression of the phosphoglycerate kinase genes of Leishmania mexicana mexicana. Molecular and Biochemical Parasitology 90:155-168.
- Anfinsen CB (1972) The formation and stabilization of protein structure. Biochem J 128: 737-749. [crossref]
- Zerrad L, Merli A, Schroder GF, Varga A, Graczer E, et al. (2011) A Spring-loaded Release Mechanism Regulates Domain Movement and Catalysis in Phosphoglycerate Kinase. J. Biol. Chem. 286: 14040-14048.
- Cheung CW, Mas MT (1996) Substrate-induced conformational changes in yeast 3phosphoglycerate kinase monitored by fluorescence of single tryptophan probes. Protein Science 5:1144-1149.
- Zomer AWM, Allert S, Chevalier N, Callens M, Opperdoes FR, et al. (1998) Purification and characterisation of the phosphoglycerate kinase isoenzymes from Trypanosoma brucei expressed in E. coli 1386: 179-188.
- Banks RD, Blake CCF, Evans PR, Haser R, Rice DW, et al. (1979) Sequence, structure and activity of phosphoglycerate kinase: a possible hinge-bending enzyme. Nature 279:773-777.
- Lijnzaad P, Berendsen HJC, Argos P (1996) A method for detecting hydrophobic patches on protein surfaces. Proteins 26:192-203.
- Teese MG, Langosch D (2015) Role of GxxxG Motifs in Transmembrane Domain Interactions. Biochemistry 54: 5125-5135. [crossref]
- Wilson MR, Kugel S, Huang J, Wilson LJ, Wloszczynski PA, et al. (2015) Structural determinants of human proton-coupled folate transporter oligomerization: role of GXXXG motifs and identification of oligomeric interfaces at transmembrane domains 3 and 6. Biochem J 469: 33-44.
- Bowler MW (2013) Conformational dynamics in phosphoglycerate kinase, an open and shut case? FEBS Lett 587: 1878-1883. [crossref]
- Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181: 223-230. [crossref]
- Gershenson A, Gierasch LM (2011) Protein folding in the cell: challenges and progress. Curr Opin Struct Biol 21: 32-41. [crossref]
- Screpanti E, Hunte C (2007) Discontinuous membrane helices in transport proteins and their correlation with function. J Struct Biol 159: 261-267. [crossref]
- Quinones W, Caceres A, Ruiz M, Concepcion J (2015) Glycosomal membrane proteins and lipids from Leishmania Mexicana. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 182: 27-36.
- Rorick MM, Wagner GP (2011) Protein structural modularity and robustness are associated with evolvability. Genome Biol Evol 3: 456-475. [crossref]
- Gupta A, Adami C (2016) Strong selection significantly increases epistatic interactions in the long-term evolution of a protein. PLOS Genetics 12: e1005960.
- Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157: 105-132. [crossref]